Ed Friedlander, M.D., Pathologist
scalpel_blade@yahoo.com



Cyberfriends: The help you're looking for is probably here.
Welcome to Ed's Pathology Notes, placed here originally for the convenience of medical students at my school. You need to check the accuracy of any information, from any source, against other credible sources. I cannot diagnose or treat over the web, I cannot comment on the health care you have already received, and these notes cannot substitute for your own doctor's care. I am good at helping people find resources and answers. If you need me, send me an E-mail at scalpel_blade@yahoo.com Your confidentiality is completely respected.
 DoctorGeorge.com is a larger, full-time service. There is also a fee site at myphysicians.com, and another at www.afraidtoask.com.
DoctorGeorge.com is a larger, full-time service. There is also a fee site at myphysicians.com, and another at www.afraidtoask.com.
Translate this page automatically
 | With one of four large boxes of "Pathguy" replies. |
 I'm still doing my best to answer
everybody.
Sometimes I get backlogged,
sometimes my E-mail crashes, and sometimes my
literature search software crashes. If you've not heard
from me in a week, post me again. I send my most
challenging questions to the medical student pathology
interest group, minus the name, but with your E-mail
where you can receive a reply.
I'm still doing my best to answer
everybody.
Sometimes I get backlogged,
sometimes my E-mail crashes, and sometimes my
literature search software crashes. If you've not heard
from me in a week, post me again. I send my most
challenging questions to the medical student pathology
interest group, minus the name, but with your E-mail
where you can receive a reply.
Numbers in {curly braces} are from the magnificent Slice of Life videodisk. No medical student should be without access to this wonderful resource. Someday you may be able to access these pictures directly from this page.
Also:
Estimating the Time of Death -- computer program right on a webpage
Freely have you received, freely give. -- Matthew 10:8. My site receives an enormous amount of traffic, and I'm
handling about 200 requests for information weekly, all
as a public service.
Pathology's modern founder,
Rudolf Virchow M.D., left a legacy
of realism and social conscience for the discipline. I am a mainstream Christian, a man of science, and a proponent of common sense and common kindness. I am an outspoken enemy of
all the make-believe and bunk that interfere with
peoples' health, reasonable freedom, and happiness. I
talk and write straight, and without apology.
Throughout these notes, I am speaking only
for myself, and not for any employer, organization,
or associate.
Special thanks to my friend and colleague,
Charles Wheeler M.D.,
pathologist and former Kansas City mayor. Thanks also
to the real Patch
Adams M.D., who wrote me encouragement when we were both beginning our unusual medical careers.
If you're a private individual who's
enjoyed this site, and want to say, "Thank you, Ed!", then what I'd like best is a contribution to the Episcopalian home for abandoned, neglected, and abused kids in Nevada:
My home page
Especially if you're looking for
information on a disease with a name
that you know, here are a couple of
great places for you to go right now
and use Medline, which will
allow you to find every relevant
current scientific publication.
You owe it to yourself to learn to
use this invaluable internet resource.
Not only will you find some information
immediately, but you'll have references
to journal articles that you can obtain
by interlibrary loan, plus the names of
the world's foremost experts and their
institutions.
Alternative (complementary) medicine has made real progress since my generally-unfavorable 1983 review linked below. If you are interested in complementary medicine, then I would urge you to visit my new
Alternative Medicine page. If you are looking for something on complementary
medicine, please go first to
the American
Association of Naturopathic Physicians.
And for your enjoyment... here are some of my old pathology exams for medical school undergraduates.
I cannot examine every claim that my correspondents
share with me. Sometimes the independent thinkers
prove to be correct, and paradigms shift as a result.
You also know that extraordinary claims require
extraordinary evidence. When a discovery proves to
square with the observable world, scientists make
reputations by confirming it, and corporations
are soon making profits from it. When a
decades-old claim by a "persecuted genius"
finds no acceptance from mainstream science,
it probably failed some basic experimental tests designed
to eliminate self-deception. If you ask me about
something like this, I will simply invite you to
do some tests yourself, perhaps as a high-school
science project. Who knows? Perhaps
it'll be you who makes the next great discovery!
Our world is full of people who have found peace, fulfillment, and friendship by suspending their own reasoning and
simply accepting a single authority that seems wise and good. I've learned that they leave the movements when, and only when, they discover they have been maliciously deceived.
In the meantime, nothing that I can say or do will
convince such people that I am a decent human being. I no longer answer my crank mail.
This site is my hobby, and I presently have no sponsor.
This page was last updated February 6, 2006.
During the ten years my site has been online, it's proved to be
one of the most popular of all internet sites for undergraduate
physician and allied-health education. It is so well-known
that I'm not worried about borrowers.
I never refuse requests from colleagues for permission to
adapt or duplicate it for their own courses... and many do.
So, fellow-teachers,
help yourselves. Don't sell it for a profit, don't use it for a bad purpose,
and at some time in your course, mention me as author and KCUMB as my institution. Drop me a note about
your successes. And special
thanks to everyone who's helped and encouraged me, and especially the
people at KCUMB
for making it possible, and my teaching assistants over the years.
Whatever you're looking for on the web, I hope you find it, here or elsewhere. Health and friendship!
{26443} platelets
QUIZBANK Hemodynamic #'s 114-197
INTRODUCTION TO THE BLEEDING DISORDERS ("HEMORRHAGIC DIATHESES") Your lecturer has no interest in presenting one more overview of the clotting cascades, the activation of platelets, or the mysteries of endothelium. We'll look today at diseases that affect one or more of these. I promise to keep it
simple. One more time... The REAL clotting cascade (i.e., how it works in life most of the time)  I am presently adding clickable links to
images in these notes. Let me know about good online
sources in addition to these:
I am presently adding clickable links to
images in these notes. Let me know about good online
sources in addition to these:
Pathology Education Instructional Resource -- U. of Alabama; includes a digital library
Houston Pathology -- loads of great pictures for student doctors
Pathopic -- Swiss site; great resource for the truly hard-core
Syracuse -- pathology cases
Walter Reed -- surgical cases
Alabama's Interactive Pathology Lab
"Companion to Big Robbins" -- very little here yet
Alberta Pathology Images --hard-core!
Cornell
Image Collection -- great site
Bristol Biomedical
Image Archive
EMBBS Clinical Photo Library
Chilean Image Bank -- General Pathology -- en Español
Chilean Image Bank -- Systemic Pathology -- en Español
Connecticut
Virtual Pathology Museum
Semmelweis U., Budapest -- enormous pathology photo collection
Iowa Skin Pathology
Loyola
Dermatology
History of Medicine -- National Library of Medicine
KU Pathology Home
Page -- friends of mine
The Medical Algorithms Project -- not so much pathology, but worth a visit
National Museum of Health & Medicine -- Armed Forces Institute of Pathology
Telmeds -- brilliant site by the medical students of Panama (Spanish language)
U of Iowa Dermatology Images
U Wash Cytogenetics Image Gallery
Urbana Atlas of Pathology -- great site
Visible Human Project at NLM
WebPath: Internet Pathology
Laboratory -- great site My team:
My team:Ed Lulo's Pathology Gallery
Bryan Lee's Pathology Museum
Dino Laporte: Pathology Museum
Tom Demark: Pathology Museum
Dan Hammoudi's Site
Claude Roofian's Site
Pathology Field Guide -- recognizing anatomic lesions, no pictures

St.
Jude's Ranch for Children
I've spent time there and they are good. Write "Thanks
Ed" on your check.
PO Box 60100
Boulder City, NV 89006--0100
More of my notes
My medical students
Clinical Queries -- PubMed from the National Institutes of Health. Take your questions here first.
HealthWorld
Yahoo! Medline lists other sites that may work well for you

We comply with the
HONcode standard for health trust worthy
information:
verify
here. 


![]()
{26169} normal megakaryocyte
{13745} megakaryocyte
{25191} hematocele (guy got kicked probably)
{39557} hemorrhage into renal pelvis (this was a TTP case)
{05938} purpura from thrombocytopenia
All diseases of inadequate hemostasis have spontaneous bleeding (petechiae, purpura, mucous membranes, GI bleeding, hematuria, into joint spaces) and/or excessive bleeding after trauma or surgery.
The range is from lethal diseases (factor VIII:C deficiency, Bernard-Soulier's, Glanzmann's) to non-diseases (factor XII deficiency, many von Willebrand's).
Three groups:
TESTING HEMOSTASIS
Platelet count: reference range is 150,000 to 400,000 (or 450,000 or 350,000);
Platelets rise at ovulation, and fall low at the start of menstruation.
{14727} finger-stick blood; platelets clumped!
Bleeding time: checks both quantity and functional quality of platelets. Usually 2-9 minutes.
Template: Use a standard spring-loaded razor device that produces a 1 mm deep, 3 mm long incision. Normally bleeds 6-10 minutes; probably this is longer than the Ivy time since platelet recruitment is slower for this tiny wound.
{14117} bleeding time, step 1
{14120} bleeding time, step 2
{14123} bleeding time, step 3
{14126} bleeding time, step 4
{14129} bleeding time, step 5
{14132} bleeding time, step 6
{14135} bleeding time, step 7
{14138} bleeding time, step 8
{14141} bleeding time, step 9
{14144} bleeding time, step 10
Thrombin time (TT): checks factor I (fibrinogen) activity
Prothrombin time (PT): checks activities of factors I, II, V, VII, and X
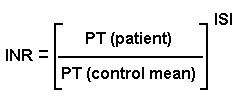
* You'll try to maintain your patients on coumarin in various ranges (2.0-3.0 to prevent deep vein thrombi in patients at high risk; 2.5-3.5 for patients with mechanical heart valves; etc., etc. Chest 108(S): 231, 1995).
Activated partial thromboplastin time (aPTT): checks activities of factors I, II, V, VIII, IX, X, XI, XII. It is prolonged when one of these drops below about 30% of healthy.
Clot retraction: checks factor I, platelet count, and a particular platelet function (missing in Glanzmann's disease)
Urea solubility: checks factor XIII ("fibrinoligase", cross-links fibrin strands, rendering the clot insoluble)
Fibrin degradation products (FDP, "fibrin split products", FSP, X-Y-D-E): increased levels suggest ongoing fibrinolysis by plasmin, particularly in disseminated intravascular coagulation (DIC); these are also likely to be elevated when a large thrombus is present.
D-dimer: From broken-down clots that have been crosslinked. Once billed as "a more specific test for DIC", it's now clear that D-dimer is elevated whenever there is intravascular thrombolysis, whether from DIC or big thrombi.
Platelet aggregation studies: testing response to ADP, thrombin, collagen, serotonin, catecholamines, and thromboxane A2 -- all "platelet agonists". Platelet function testing explained: Arch. Path. Lab. Med. 126: 133, 2002. Remarkably accurate new assay: Am. J. Clin. Path. 122: 178, 2004.
Mixing studies: Mix the patient's blood half-and-half with blood from somebody who is healthy. If the patient has an antibody against a clotting factor, the tests will remain abnormal. If the patient is missing something important, the tests will become normal.
Tip: Get a functional, rather than an immune-based, assay for proteins C, S, AT-III, etc., etc. Some people's factors may contain a mutation that renders them non-functional but still demonstrable by immune methods.
CD40 ligand: Platelet protein; up-and-coming test to show that there's platelet activation in the body, i.e., this patient's chest pain is likely to turn into an infarct (NEJM 348: 1104, 2003).
INCREASED VASCULAR FRAGILITY ("nonthrombocytopenic purpuras", etc.): bleeding problems despite normal platelet count, bleeding time, PT, aPTT, TT, FDP)
Bleeding from fragile vessels, with normal platelets and coagulation, is seldom serious.
Massive bleeding is rare. Vascular fragility is more likely to cause skin bruises, dependent petechiae, gum bleeding (eating, toothbrushing), hematuria, nosebleeds, GI bleeds.
Causes include:
infections (meningococcemia, gonoccemia, scarlet fever, SBE, other forms of sepsis, rickettsial disease, the many viral hemorrhagic fevers, Ebola being only one) that particularly damage endothelium
amyloidosis
collagen problems: scurvy, Ehlers-Danlos syndrome ("the human pretzel"), Cushing's syndrome, osteogenesis imperfecta (patients do not convert reticulin to regular collagen normally), hereditary hemorrhagic telangiectasis (Osler-Weber-Rendu disease; patients have an abnormal type III collagen or some related mutation and some vascular malformations that bleed easily), "old age" ("senile purpura")
{05940} scurvy, gums
{38195} scurvy case, bone
immune-complex deposition in vessel walls: serum sickness, Henoch-Schonlein purpura, cryoglobulinemia, wonder drugs (the usual cause of "leukocytoclastic vasculitis", lots of living and dead neutrophils around the cutaneous vessels, which in turn is the common cause of "palpable purpura" on the skin)
{12261} erythema multiforme case with purpura
{12262} erythema multiforme case with purpura
{12529} erythema multiforme case with purpura
* mind phenomena (Gardner-Diamond autoerythrocyte sensitization / psychogenic purpura, religious stigmatization; do you believe in this stuff? I don't know....)
As noted, the usual platelet and coagulation tests are normal. (Capillary fragility tests using blood pressure cuffs, the old "tourniquet test", etc., are not very useful.) REDUCED PLATELET NUMBER ("THROMBOCYTOPENIA"): Ped. Clin. N.A. 51: 1109, 2004, lots more
Platelets are the first line of defense against bleeding, plugging up little holes in capillaries (primary hemostasis). Of course, they also help initiate coagulation (to solidify the blood before it gets through the bigger
holes), and eventually become an important component of most clots. Think of platelets as little band-aids that turn into bricks (fibrin's the mortar). Excessive bleeding due to platelet disorders is apparent almost immediately after trauma. Excessive bleeding due to coagulation factor
deficiency becomes apparent only after a few minutes. Thrombopoietin ("megapoietin") finally cloned and characterized: Nature 369: 533, 1994; Proc. Nat. Acad. Sci. 91: 11104, 1994; Proc. Nat. Acad. Sci. 94:
4669, 1997. * Thrombopoietin is now being used to make people make extra platelets prior to chemotherapy; these are harvested and re-transfused (Lancet
359: 2145, 2002).
Here's how the feedback works: Something in the body produces a constant amount of thrombopoietin, and most of it gets sopped up by the circulating platelets. Any that's left-over stimulates the production of more platelets.
* Thrombin does some carving on thrombopoietin and perhaps this is why more platelets are produced when you start clotting. Stay tuned.
Always order a CBC, which includes
a platelet count, on anybody who seems seriously sick. Thrombocytopenia is said to be present when platelet count is less than 100,000/mcL. Platelet problems are often heralded by petechiae on the skin and mucosal surfaces. Bleeding after trauma (surgery, etc.) can be a problem
when platelet count is below 40,000/mcL. Spontaneous bleeding likely to occur only when the platelet count is below 20,000 or so. (Petechiae and purpura randomly over the skin, blood blisters in the mouth, GI, GU, CNS bleeding.)
{11526} hemorrhage in thrombocytopenia (* "Sweet's syndrome" case) Severe spontaneous bleeding may be expected when count gets below 10,000/mcL. Causes of thrombocytopenia are many.
{13805} giant platelets, myelofibrosis case
Tip: If you see only a few platelets and many of them are large, the patient is probably turning out platelets very rapidly (i.e., they have not fully separated from one another), i.e., they are being destroyed peripherally. Of
course, to be sure, you may want to do a bone marrow exam. If platelets are being destroyed peripherally, you will see many, young megakaryocytes with hypo-segmented nuclei.
* Pitfall: When remission of a leukemia is being induced, the fragmentation of the white cells may result in fragments that
are interpreted by the automated cell counters as platelets, disguising a serious thrombocytopenia (Arch. Path.
Lab. Med. 123 1111, 1999).
Patients with platelet abnormalities have increased bleeding time, normal PT, aPTT. To distinguish a thrombocytopenia of decreased production (few or non-maturing megakaryocytes) or increased destruction (increased megakaryocytes), examine the bone marrow. If platelets are rapidly being destroyed and the marrow
is making them overtime, giant platelets are often abundant in the peripheral blood.
Isoimmune thrombocytopenia
Neonatal: Fetal-maternal incompatibility, analogous to hemolytic disease of the newborn. Mother's IgG antibodies against some specific platelet antigen on baby's platelets causes them to be destroyed. Read all about it: NEJM 337: 32, 1997. Post-transfusion: After a PlA1-negative patient receives someone else's PlA1-positive platelets (to which the patient must be already sensitized), she starts destroying her own platelets. (Immune complexes adsorbed to the
patient's platelets are probably the cause.) Idiopathic thrombocytopenia purpura ("ITP", autoimmune thrombocytopenic purpura, "ATP", etc): Acute ITP: a disease of children, most often following a viral infection. Platelets become coated with antibodies (probably not anti-platelet autoantibodies, but immune complexes from the viral illness) and get eaten by the RE system. Thrombocytopenia in AIDS results from this mechanism, and/or
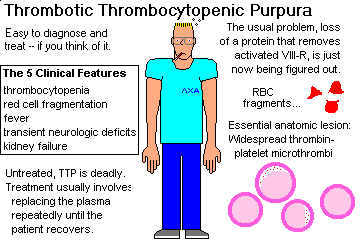
autoantibodies against IIb-III and/or HIV attacking megakaryocytes that are positive for CD4 Chronic ITP: a disease of adults, especially those with autoimmune disease. True anti-platelet antibodies are present, and platelets are destroyed by the RE system as in acute ITP. Both types have increased IgG in the platelet fraction. Splenectomy is necessary in some cases. (The spleen makes much of the offending antibody, and it eats most of the sensitized platelets.) Thrombotic thrombocytopenic purpura (TTP; review Am. J. Clin. Path. 121 S: S-89. 2004)
This is a dread disease that kills young adults. It is characterized by thrombocytopenia, microangiopathic hemolytic anemia (i.e., DIC), fever, transient neurologic defects, and renal failure.
Microthrombi occur in the arterioles and capillaries of most organs.
They're almost always worst in the heard.
They are made of loose aggregates of platelets, von Willebrand's factor, and some fibrin.
The pathophysiology has finally yielded up its secrets with the
discovery of von Willebrand factor cleaving protease (which removes big aggregates of von Willebrand's factor from the blood).
Familial TTP features a familial deficiency of this protease
(NEJM 339:1578, 1998; gene ADAMTS-13 Nature 413: 438, 2001; Blood 102: 1148, 2003).
TTP may be secondary to lupus (must be the autoantibody...), HIV-infection (South. Med. J.
88: 82,
1995), * ticlopidine (as after coronary stenting, JAMA 281: 806, 1999)
and others.
Today you may diagnose it by the finding of severe thrombocytopenia plus microangiopathic
hemolysis without alternative explanation; your pathologist can help by finding
big aggregates ("ultralarge multimers") of vWF in the blood (Am. J. Clin. Path. 121-S: S89, 2004).
Once uniformly fatal, the disease is now being cured. The treatment involves administration of fresh-frozen plasma (Lancet 345: 224, 1995), up to complete plasma exchange. {08059} petechiae on heart, leukemia case DEFECTIVE PLATELET FUNCTION (normal platelet count, prolonged bleeding time)
What platelets do for you:
Step 1: Adhere (i.e., to collagen and basement membrane)
Step 2: Release their ADP (makes platelets work better) and their catecholamine (local vasoconstriction) and their thrombin (activates fibrin directly)
Step 3: Aggregate (i.e., stick tight and recruit more platelets, due to ADP) Step 4: Fuse tight to fibrin and each other, to seal the leak Step 5: Retract (i.e., pull the clot tight)
Note that as long as platelet release and fibrin production are reasonably normal, there will not be bleeding from small nicks (i.e., shaving) even in patients with hemophilia. * Lab workup of platelet disorders: Arch. Path. Lab.
Med, 126: 133, 2002. Hereditary:
Defects of platelet adhesion
Bernard-Soulier disease ("giant platelets syndrome")
An autosomal recessive, severe bleeding disorder.
Giant, useless platelets that won't stick to the subendothelium; severe bleeding.
There is a deficiency of certain glycoproteins (* notably Ib-IX)
that bind the factor (VIII:R) that enables platelets to interact to collagen.
Stem cell transplantation for cure: Ann. Int. Med. 138: 79, 2003.
There is a mild dominant allele (big platelets, lowish counts): Blood 97: 1330, 2001;
Am. J. Med. 112: 742, 2002.
Von Willebrand's disease (see also below): Review Medicine 76: 1, 1997. An autosomally inherited, qualitative or quantitative lack of Von Willebrand's factor. There is no aggregation in response to ristocetin, but there is normal aggregation in response to epinephrine, collagen, ADP.
The platelet defect is corrected by normal or hemophilic ("factor VIII-deficient") plasma.
Defects of platelet secretion (of prostaglandins and ADP, the "release reaction"): various "aspirin-like" hereditary diseases too rare and complicated to outline here! * Storage-pool disease. Leave this arcane stuff to the hematologists.
* Hermansky-Pudlak syndrome: Any of several genetic diseases with defective production
of melanosomes, platelet granules, and lysosomes (Blood 96: 4227, 2000); patients are albinos
* Gray-platelet disease: No granules. Autosomal-variable, and a bleeding problem. Thankfully
rare. See Blood 98: 1382, 2001.
Defects of platelet aggregation
Thrombasthenia (Glanzmann's disease, formerly "tired platelet syndrome"): An uncommon, autosomal recessive, severe bleeding disease.
Platelets fail to aggregate on an un-anticoagulated peripheral smear,
with ADP, collagen, epinephrine, or thrombin. Clot retraction is also absent.
(They lack the
integrin glycoprotein GPIIb-IIIa that binds
that fibrinogen that in turn bridges platelets. Read all about it: Am. J. Hum. Genet. 53: 140, 1993; molecular biology Blood 90: 669, 1997.)
Acquired defects:
Aspirin permanently inhibits
cyclooxygenase (by acetylating it), preventing production
of thromboxane A2 and
producing an acquired secretion defect that lasts for the life of the platelet (a platelet lives about 9 days)
The other NSAIDS temporarily block cyclo-oxygenase.
* All about NSAIDS and platelets: Am. J. Med. 106 (5B): 25S, 1999. In uremia, there is a complex platelet defect reversed by dialysis, which may be due to * phenol and/or * guanidinosuccinic acid.
* Alcohol enhances the effect of aspirin on platelets. During an alcoholic binge, the platelet numbers drop; during alcohol withdrawal, there is a rebound thrombocytosis that can produce a stroke if somebody is predisposed.
THROMBOCYTOSIS
Thrombocytosis is a platelet count above 400,000/mcL. It may occur as a rebound after severe bleeding, after surgery or sepsis,
following splenectomy, or just runs in the family, or is part of the disease in iron deficiency (common, but no one knows why), carcinomatosis, or * Hodgkin's disease. Thrombocythemia is a sustained platelet elevation over 800,000/mcL.
Unless there's some other obvious explanation,
this indicates some myeloproliferative disorder (leukemia, polycythemia vera, early myelofibrosis, etc). If there is no other known illness, it is called "essential thrombocythemia", a myeloproliferative disorder, and the
patient may have either thrombi or platelet insufficiency.
Today's criteria allow the diagnosis if the platelet count is consistently
over 600,000 and a workup (including marrow tap) shows no cause (especially, no bcr/abl):
Ann. Int. Med. 139: 470, 2003. This will soon be replaced by
clonality assays (Blood 101: 3294, 2003).
Even then, it's likely to remain
asymptomatic for a long time,
and is seldom lethal (Am. J. Med. 117: 755, 2004).
Update Mayo Clin. Proc. 80: 97, 2005. (* New remedy
works wonders: anagrelide. Many current references. Hydroxyurea plus
low-dose aspirin may work even better: NEJM 2005: 353, 2005.)
{24786} essential thrombocythemia
ABNORMALITIES IN COAGULATION FACTORS (abnormal PT, aPTT, TT, and/or FDP; usually normal platelet count and bleeding time)
Deficiencies of all of the clotting factors have been described. For each, the deficiency can be mild or severe, the protein can be absent or just defective, or the deficiency may be due to an inhibitor (antibody, etc.)
against it. Deficiencies may be hereditary or acquired:
Hereditary deficiencies involve a deficiency of a single factor. Acquired deficiencies (except those due to an autoantibody) involve several factors Deficiency of vitamin-K-dependent factors (neonates, malabsorption, heavy antibiotics, coumarin therapy, bad liver trouble)
Remember the body requires vitamin K so that the liver can make gamma-carboxyglutamic acid, present only in factors II, VII, IX, and X.
* Of these, factor VII is the first to go, so the defect will appear initially in the extrinsic pathway, i.e., abnormal PT.
Heparin therapy potentiates antithrombin III, so heparin indirectly inactivates thrombin. * The thrombin time estimate for fibrinogen is useless in the heparinized patient, so the lab uses snake venom ("Russell viper venom time, RVVT") instead of thrombin. "Circulating anticoagulant" is the common term for any abnormal protein, not part of the normal clotting-anti-clotting systems, that interferes with coagulation. Such a protein may be either an autoantibody against a clotting factor, or an inhibitor of one or more steps. Both are common enough in systemic lupus, rare in other people. Disseminated intravascular coagulation (all factors consumed, as are platelets.) Patients with coagulation factor deficiencies rarely have spontaneous petechiae or purpura. Instead, they get ecchymoses or hematomas after minor injury that the platelets don't handle. Bleeding for days after tooth extractions,
or bleeding into joint spaces (hemarthroses) are common. DEFICIENCIES OF FACTOR VIII COMPLEX
Factor VIII:C (procoagulant) is the clotting factor required to activate factor X in the intrinsic pathway. It is coded on X-chromosome. It circulates bound to VIII:R (von Willebrand's factor, made in endothelium and
megakaryocytes) which is required for the interaction of platelets with subendothelial collagen and also protects VIII:C from destruction.
VIII:R is required for platelet aggregation by ristocetin. Von W's factor is a tumor marker for Kaposi's sarcoma (of endothelial origin) and other angiosarcomas.
Sex-linked recessive deficiency of factor VIII:C.
This is a mild, moderate, or severe bleeding disorder affecting 1 in every 5000 men. Female carriers may have mild disease due to unlucky lyonization (Blood 85: 599, 1995). Severe cases have maybe 1% or less activity of the healthy protein. Hemarthroses, especially in the knees, are excruciatingly painful and
lead to crippling early in life. No one knows why bleeding in the knees and
other big weight-bearing joints is so
common in the hemophilias.
At least in the developed nations, most of these men now lead near-normal lives
thanks to recombinant factor VIII.
* Managing these patients
is expensive
but works; keys are activated factor VII (Blood 102: 2358, 2003)
and inducing tolerance (Blood 96: 1698, 2000). Gene therapy for hemophilia should be routine in the next decade
or so. It has already
worked in animals, and a phase 1 study (UC Davis) has some hopeful results
(Sept. 2003: Blood 102: 2038, 2003).
An earlier attempt using fibroblasts,
without even a virus, raised levels;
the trick is to get the fibroblasts to continue making the factor VIII for more than a few months(NEJM 314: 1735, 2001).
VON WILLEBRAND'S DISEASE ("pseudohemophilia")
Probably the commonest inherited hemorrhagic disorder.
It involves a qualitative or quantitative deficiency of
VIII:R/vWF (thus often also low VIII:C as it's not protected)
and/or the platelet factor to which it binds
(* glycoprotein Ib α).
Thus there is prolonged bleeding time and, in severe cases, some prolongation of aPTT.
And of course the platelets don't stuck together well; in particular, they
fail to respond to ristocetin, which should active the vWF receptors on
platelets )
resulting in the vWF gluing the platelets together. If you don't understand this, please review it.
Autosomal inheritance varies according to subtype: dominant (asymptomatic to moderate forms) or recessive (severe forms; carriers are asymptomatic).
All the von Willebrands' syndromes are autosomal dominant and relatively mild,
with the exception of type III, which results from two doses.
Easy...
II. Mutant vWF
III. Two doses, severe illness
In a woman with heavy periods and a normal pelvic exam, von Willebrand's is quite likely: Lancet 351: 485, 1998. Remember that von Willebrand's factor is an acute phase reactant, so levels may be normal
during other illnesses. Sex hormones (especially estrogens) partially correct the molecular deficiency in some types, so the disease often gets better at puberty.
The management of von Willebrand's disease, especially before surgery,
used to be based on administering factor VIII concentrates, which are full
of good vWF multimers. Treatment was revolutionized in the 1980's by the discovery that
desmopressin (!) raises vWF levels.
Acquired von-Willebrand's has at least three etiologies.
Think also of autoantibodies, and adsorption to the surfaces of tumor cells
(Mayo Clin. Proc. 77: 181, 2002).
FACTOR IX DEFICIENCY (hemophilia B, Christmas disease) Sex-linked recessive deficiency, often even more severe than, but otherwise similar to, classic hemophilia. One man in 25,000 is affected. Again, the joint disease is the most troublesome feature from day to day.
Mr. Christmas was the first patient discovered to have factor IX deficiency.
Like patients with factor VIII deficiency, most of these people now enjoy near-normal lives,
the recombinant protein having been introduced in 1999. Only about 3% make troublesome antibodies against factor IX.
* Attempts at cure with gene therapy are of course ongoing. The trick is
getting the transfected cells to pump the stuff out (Blood 101: 2963, 2003).
* Alpha-2 plasmin inhibitor (antiplasmin) deficiency has been described; you remember this is the stuff that binds up any active plasmin that makes it into the flowing blood (Br. J. Hem. 114: 4, 2001). The treatment is to
administer tranexamic acid or epsilon-amino caproic acid, both of which prevent binding of plasminogen to fibrin.
Lots more.
HYPERCOAGULABLE BLOOD: "Thrombophilia"; "hypercoagulopathy". Not rare,
but tends to get overlooked.
Big reviews: Am. J. Med. 116: 81, 2004;
Ann. Int. Med. 138: 128, 2003;
Postgrad. Med. 101(5): 249, May 1997.
Lab screening: Am. J. Clin. Path. 108: 434, 1997.
Acquired:
* Prothrombin G20210A is a point mutation affecting 2% of people; it renders blood
slightly more coagulable. It is insufficient, by itself, to cause
thrombosis. It can be detected only by DNA testing and
only recently have people started talking about this
being worthwhile (Arch. Path. Lab. Med. 126: 1319, 2002,
contrast Mayo Clin. Proc. 75: 595, 2000). Protein C
deficiency (Blood 85: 2756, 1995) and protein S deficiency are
relatively common; 1 person in 300 is heterozygous for lack of protein C. (The most severely affected homozygotes get lethal purpura
fulminans as babies.) You know that thrombomodulin on intact endothelium activates protein C (why is that good?), and that S and C work together to destroy Va and VIIIa. These patients (notably the homozygotes, protein C deficient
heterozygotes are at around 8x increased risk, homozygotes 80x: Lancet 341: 134, 1993) clot their blood too readily, and are prone to pulmonary emboli and so forth. Both protein S and protein C are vitamin K dependent
proteins. Thus the benefits of warfarin therapy are probably limited; this is recently supported (Arch. Int. Med. 157: 2227, 1997); since warfarin depresses protein C levels before it depresses II, VII, IX, and X, these people
may actually develop skin necrosis from taking the medication; see Ob. Gyn. 90: 671, 1997; Br. J. Surg. 87: 266, 2000.
Hereditary deficiency of antithrombin III is quite common. When it's just a matter of too little being produced (* type I AT3
deficiency), there's an increased risk especially for deep vein thrombi. When it's mutated (* type II AT3 deficiency), perhaps the patient will not respond to heparin (which works by enhancing the effect of normal AT3). We've already
probed the mysteries of antiphospholipid antibody syndrome (Blood 86: 617, 1995; Am. J. Med. 100: 530, 1996; mega-review Lancet 353: 1348, 1999; Arch. Path. Lab. Med. 126: 1326, 2002).
Two different types of antiphospholipid antibodies are described:
"anticardiolipin antibody", common, especially serious if longstanding and IgG; a simple ELISA assay makes the diagnosis
Should you anticoagulate them
all (NEJM 332: 993, 1995)? Give them low-dose aspirin? Do nothing unless they're very sick (Am. J. Ob. Gyn. 176: 1099, 1997)? Nobody knows yet what's best.
Right now, it seems that the antibodies activate endothelial cells (Circulation 99: 1997, 1999).
we're discovering that
many of them also make autoantibodies against proteins C and/or S, and the autoantibody itself may induce TF on the surfaces of monocytes, etc. (Lancet 350: 1491, 1997).
"Activated protein C resistance" is usually caused by a particular point mutation of factor V (V Leiden). It is a common, infamous cause of thrombosis, pulmonary emboli
(NEJM 336: 399, 1997; about half of young folks with "unexplained" DVT's have it), second-trimester miscarriage / preterm birth (Lancet 358: 1238, 2001), and accelerated atherosclerosis (NEJM 332: 912, 1995).
This factor V resists the
anticoagulant effect of protein C (J. Lab. Clin. Med. 125: 566, 1995). This was recognized in 1995 as the most common of the then-five known major hypercoagulability syndromes, affecting maybe 3% of the public.
A major 1998 study found no benefit from long-term
anticoagulation of V-Leiden people (BMJ 316: 95, 1998.)
Not surprisingly, V-Leiden is a significant coronary risk factor: Am. Heart J. 147: 897, 2004.
Stay tuned: Hyperhomocysteinemia resulting from any of several kinks in methionine metabolism, or perhaps even just lack of folate in the diet (stay tuned),
is now known to be a serious risk factor both
for accelerated atherosclerosis and venous thrombi. Update Arch. Path. Lab. Med. 126: 1367, 2002.
Homocysteine damages the endothelium, and does various things to various coagulation factors that are presently being worked out.
Mild forms may be extremely common (Lancet
345: 902, 1995; NEJM 334: 759, 1996; Am. J. Clin. Path. 108: 115, 1997). fortunately, you can treat it with vitamin B12 and folic acid.
* Lupus anticoagulant, factor V Leiden, prothrombin G20210A, and protein S deficiency
seem to place a woman at increase risk for fetal loss: Lancet 361: 901, 2003.
A major piece of news is that the hereditary thrombophilias are serious risks for clots in the spiral and intervillous arteries of the pregnant uterus. This in turn places the pregnancy at risk for severe pre-eclampsia, abruption,
fetal growth retardation, and stillbirth (NEJM 340: 9, 1999.)
Uh... Please don't work up thrombophilia immediately after the acute episode. The labs will be off (why?)
DISSEMINATED INTRAVASCULAR COAGULATION ("DIC", defibrination syndrome; "death
is coming")
We have already reviewed this in "general pathology".
This is an acquired deficiency of clotting factors and platelets; they are being used up. RBC's also get shredded on the intravascular strands, producing helmet cells, schistocytes, blister cells, keratocytes, etc.
The management of disseminated intravascular coagulation has been revolutionized
by the introduction of activated protein C (Br. Med. J. 327: 974, 2003).
DIC may be caused by:
release of thromboplastin into the bloodstream obstetrical catastrophe (thromboplastin from placenta or amniotic fluid embolism)
major tissue injury (burns, heat stroke, surgery, trauma) acute promyelocytic leukemia (thromboplastin from "pro"'s granules) mucinous adenocarcinomas (mucin
activates factor X directly) sepsis (thromboplastin from PMN's)
* filovirus infection (Ebola, Marburg) -- monocytes express tissue factor on their surfaces Lancet Inf. Dis. 4: 487, 2004.
snakebite
endothelial damage
vasculitis (especially malignant hypertension, meningococcemia, rickettsial disease) Kasabach-Meritt syndrome (giant hemangioma with ongoing "localized DIC" inside) hypothermia (as in cardiac surgery: poorly understood. Sem.
Thorac. Card. Surg. 9: 246, 1997.)
"Chronic DIC" ("compensated DIC") may result from underlying malignancies, myelodysplastic syndromes, * PNH, or "idiopathic". The body may overcompensate by increasing the platelet numbers above normal, but the clotting factors remain
relatively depleted.
Treatment of DIC is that of the underlying disease.
{03178} DIC, kidney, gross with hemorrhages Remember:
Defects of the extrinsic pathway (normal aPTT, prolonged PT) usually indicate early liver disease or coumarin therapy (congenital factor VII deficiency is rare)
Defects of the intrinsic pathway (normal PT, prolonged aPTT) include factor VIII and IX deficiencies or circulating anticoagulants (congenital factor XI and XII deficiencies are rare) Defects of both pathways (prolonged PT and
aPTT): usually indicate heparin or coumarin therapy, advanced liver disease, or circulating anticoagulants (congenital factor II, V, and X deficiencies are rare)
Defects of neither pathway (normal PT and aPTT): fragile vessels, platelet problem, or factor XIII deficiency (remember urea solubility test)
Cryoprecipitate contains fibrinogen, vWF, factor VIII, factor XIII, and fibronectin, but no factor IX.
POSTSCRIPT: "INTELLIGENT DESIGN" AND "IRREDUCIBLE COMPLEXITY"
A few of the early Christian writers argued that the earth could
not be round. They used several fallacies, most famously
that people on the other side would fall off and that there would be nothing
to hold it up.
These writers claimed that all non-Christian thought was
deeply flawed and led to immorality and the world's evils.
The great church leader Augustine, who is probably best-known today
for his "Confessions", urged these people to stop. Every informed
person knew they were wrong -- ludicrously so -- and they were discrediting the Christian faith.
During the middle ages, Anselm, one of the archbishops of Canterbury,
came up with the "ontological argument" to prove the existence of God.
By definition nothing can be better than God, existing is better than not
existing, so God must exist. I doubt that many people have ever found this very
persuasive. Some of Anselm's own contemporaries didn't,
and Anselm replied graciously to those who disagreed.
From what I have read and heard,
the clotting cascade is the centerpiece of today's most-often-cited proof
of the existence of God. The claim that every portion of the clotting cascade
is finely-tuned to work together was popularized by Michael B---,
the only "intelligent design" advocate at the national level with bona fide scientific
credentials. (I refuse to recognize the one other guy, whose scientific
training was sponsored -- with the intent that he would write creationist
books -- by a cult that has taught that its founder conceived his eight children
by blowing in his wife's ear. The other three major players are all
attorneys, who are professionally trained to argue positions
even when they know they are wrong.) Unlike
Archbishop Anselm, the intelligent design
proponents (though superficially polite)
accuse their opponents of the vilest motives
and of rank stupidity.
Real scientists will recognize the old creationist
fallacy, "How could A evolve without
B, and how could B evolve without A?" Of course, things evolve together.
And B---'s claim that the coagulation cascade is "irreducibly complex"
is indisputably false.
The evolution of the clotting cascade is well-documented,
and as Darwin's theory predicts, it seems to give the same
phylogenetic tree as classic comparative anatomy. (If this really failed
for even a
single protein or gene, Darwin's theory would be refuted and "intelligent design"
pretty much established.
Are people like B--- looking?
Of course not.)
Whales lack factor XII, the gene being inactivated.
Turtles lack factors XI and XII.
Fish lack prekallikrein, XI, and XII.
Lampreys have a primitive system with tissue factor, prothrombin,
and fibrinogen.
Obviously the rest of the cascade
developed unit by unit to modulate the primitive system.
And this totally refutes the idea of "irreducible complexity."
See PNAS 100: 7257, 2003.
Origins of the vertebrate coagulation system: Thromb. Hemo. 89: 420, 2003
(England); Blood Cell. Mol. Dis. 29: 57, 2002.
There's a review of all the vertebrate systems
in J. Thromb. Hemo. 1: 1487, 2003.
Conservation back to the horseshoe crab: J. Mol. Bio. 282:
459, 1998.
The common origin of the clotting and complement cascades
(like Darwin's finches, everything used to do something else):
Trend Bioch. Sci. 27: 67, 2002.
A theologically-inclined guy at Harvard reviews
this in J. Thromb. Hemo. 1: 227, 2003.
It is inconceivable that the "intelligent design" proponents
do not know this
by now, and the fact that they persist tells me a great deal about
who they really are. If you are involved with this stuff, please stop.
If you (like me) are a person of faith, you should demand that people
stop spreading lies as "proof of the existence of God."
I find nothing "spiritual" or "moral"
about wholesale breaking of the ninth commandment.
The Christian Bible compares our present "animal bodies" to
our future state as spiritual beings.
Like Job, I prefer to stand in awe and not demand answers to everything
right now. I expect you do, too.
 Henoch-Schonlein purpura
Henoch-Schonlein purpura
mdchoice.com
Gardner-Diamond ("autoerythrocyte sensitization", "psychic purpura")
is the strangest of the lot -- platelet aggregation
studies are abnormal, and painful bruises appear around the body.
Unlike other folks, te lesions are reproduced by intradermal injection of
the patient's own washed red cells. It responds to removal of the stressors.
See Eur. J. Haem. 65: 144, 2000.
Your lab can now assay thrombopoietin: Eur. J. Hem. 61:119, 1998.
This dread "white clot disease"
is caused by autoantibodies against a complex of heparin and platelet factor 4 (Blood 94: 208, 1999); we are seeing less nowadays thanks to the less-immunogenic low-MW heparin;
update Arch. Path. Lab. Med. 126: 1415, 2002
Chronic ITP tends to be mild and
presents only a small risk to life. Some physicians do not treat unless counts fall below 30,000 (Lancet 359: 4, 2002).
* Future pathologists: Want to
know if the surgeon got the entire spleen, and didn't leave an accessory spleen or a bit of spilled spleen tissue behind? Fully-splenectomized patients have Howell-Jolly bodies in their circulating red cells!
People with acquired TTP have an autoantibody against this protease (NEJM 339: 1585, 1998).
Hemolytic-uremic syndrome is a kidney-only microangiopathy in which thrombocytopenia is less prominent.
Compare-and-contrast with TTP: Arch. Path. Lab. Med. 127: 834, 2003.
More about this under Kidney.
{21433} petechiae from football mouth-guard, no hemostasis problem
 Pseudo-Gray Platelets
Pseudo-Gray Platelets
Virginia
Good pictures
* Future cardiologists: This is the same glycoprotein complex that abciximab therapy is directed against in acute coronary disease: Am. Heart J. 138: S16 & S24, 1999.
* {13748} "megakaryocytic myelosis"
 CLASSIC HEMOPHILIA ("factor VIII deficiency", hemophilia A, "royal blood"): Review of the two major hemophilias:
Lancet 361: 1801, 2003.
CLASSIC HEMOPHILIA ("factor VIII deficiency", hemophilia A, "royal blood"): Review of the two major hemophilias:
Lancet 361: 1801, 2003.
Fewer than 10% of patients are ever troubled by autoantibodies against factor VIII,
but when it does happen, it can be a disaster. Why?
* Hemophilia in crown prince Alexis enabled the charlatan Rasputin
to gain much control of the Russian royal family, a disaster that helped lead
to the Bolshevik revolution. Two modern physicians examine what really
happened: Am. J. Surg. 145: 193, 1983 (great read). Hemophilia in the
royal families of Europe: Br. J. Haem. 105: 25, 1999.
I. Enough vWF is not made. This is by far the most common.
At least half these people are
completely asymptomatic (Blood 101: 2089, 2003)
IIa. Failure to cleave multimers, so the vWF binds poorly to platelets)
IIb. Large complexes bind inappropriately to platelets, which are then cleared; thrombocytopenia; increased reactivity to ristocetin; don't use desmopressin (why not?)
IIn. Binds to platelets much better than to VIII; low VIII levels
It is common in the presence of aortic valve stenosis (!) The shear
forces somehow damage the factor multimers so that they don't stick well to collagen or
platelets, even though they still bind well to factor VIII. NEJM 349:
343, 2003.
MORE HEREDITARY BLEEDING DISORDERS
* Factor V deficiency: parahemophilia.
Remember these causes:
Hereditary: As you'd expect, all except hereditary hyperhomocysteinemia are autosomal dominant, with double-dose being more severe. This list is most-common to least-common.
This features:
"lupuslike anticoagulants", relatively uncommon; specific assays exist but are tricky, and most often it'll be diagnosed presumptively by finding that adding extra phospholipid returns the clotting time to normal (* future pathologists:
use diluted russell viper venom);
Indeed, we still don't know why people with antiphospholipid antibody get hypercoagulable blood in the first place.
 Antiphospholipid antibody
Antiphospholipid antibody
Lost baby
Pittsburgh Pathology Cases
* Future pathologists: You'll diagnose this using PCR and/or discovering that adding activated protein C to the tubes doesn't double the PTT. Assays
Arch. Path. Lab. Med. 126: 577, 2002.
You are already familiar with hypercoagulable blood as a complication of cancer, notably adenocarcinomas, notably those of the pancreas ("Trousseau's other sign"). A full cancer workup is probably NOT worthwhile after an episode of
primary deep vein thrombosis: NEJM 338: 1169, 1998.
The definitive cause of Trousseau's remains to be discovered.
"Cancer procoagulant A", described a decade ago as a cysteine protease, still awaits definitive
characterization
(Arch. Bioch. 428: 131, 2004).
 Kasabach-Merritt syndrome
Kasabach-Merritt syndrome
Pittsburgh Pathology Cases
{03180} DIC, glomerulus, with fibrin-platelet thrombi
{03189} DIC, petechiae on heart
{03192} DIC, liver with recent infarcts
{09629} DIC, fibrin-platelet thrombus in brain
{39649} DIC, fibrin-platelet thrombi
{39817} schistocytes
{13895} schistocytes
{12237} Kasabach-Merritt syndrome patient
Factor VII deficiency
Pittsburgh Pathology Cases

Nov. 4, 2005: The principal "intelligent design"
website (the D--- I---) mentions the business about clotting in
lampreys, and provides a link
to a page by B--- on which the data is supposedly reviewed and the
argument is supposedly refuted. But the linked page doesn't even mention it.
You can find it yourself. This is typical of how disinformation artists operate.

Visitors to www.pathguy.com
reset Jan. 30, 2005:
Ed says, "This world would be a sorry place if
people like me who call ourselves Christians
didn't try to act as good as
other
good people
."
Prayer Request
 Drop by
and meet Ed
Drop by
and meet Ed

 Teaching Pathology
Teaching Pathology
PathMax -- Shawn E. Cowper MD's
pathology education links
Ed's Autopsy Page
Notes for Good Lecturers
Small Group Teaching
Socratic
Teaching
Preventing "F"'s
Classroom Control
"I Hate Histology!"
Ed's Physiology Challenge
Pathology Identification
Keys ("Kansas City Field Guide to Pathology")
Ed's Basic Science
Trivia Quiz -- have a chuckle!
Rudolf
Virchow on Pathology Education -- humor
Curriculum Position Paper -- humor
The Pathology Blues
 Ed's Pathology Review for USMLE I
Ed's Pathology Review for USMLE I
![]()
![]()

Pathological Chess

Taser Video
83.4 MB
7:26 min

 Crookston Collection
Crookston Collection
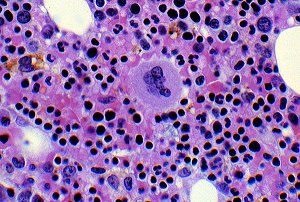 Megakaryocyte
Megakaryocyte Rare Bleeding Disorder Database
Rare Bleeding Disorder Database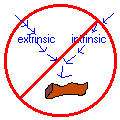
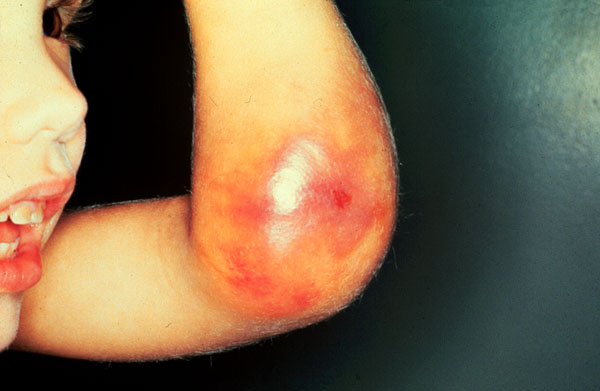 Hemophilia
Hemophilia DIC in a baby, etc.
DIC in a baby, etc.