Ed Friedlander, M.D., Pathologist
scalpel_blade@yahoo.com



Cyberfriends: The help you're looking for is probably here.
Welcome to Ed's Pathology Notes, placed here originally for the convenience of medical students at my school. You need to check the accuracy of any information, from any source, against other credible sources. I cannot diagnose or treat over the web, I cannot comment on the health care you have already received, and these notes cannot substitute for your own doctor's care. I am good at helping people find resources and answers. If you need me, send me an E-mail at scalpel_blade@yahoo.com Your confidentiality is completely respected.
 DoctorGeorge.com is a larger, full-time service.
There is also a fee site at myphysicians.com,
and another at www.afraidtoask.com.
DoctorGeorge.com is a larger, full-time service.
There is also a fee site at myphysicians.com,
and another at www.afraidtoask.com.
Translate this page automatically
 |
With one of four large boxes of "Pathguy" replies. |
 I'm still doing my best to answer
everybody.
Sometimes I get backlogged,
sometimes my E-mail crashes, and sometimes my
literature search software crashes. If you've not heard
from me in a week, post me again. I send my most
challenging questions to the medical student pathology
interest group, minus the name, but with your E-mail
where you can receive a reply.
I'm still doing my best to answer
everybody.
Sometimes I get backlogged,
sometimes my E-mail crashes, and sometimes my
literature search software crashes. If you've not heard
from me in a week, post me again. I send my most
challenging questions to the medical student pathology
interest group, minus the name, but with your E-mail
where you can receive a reply.
Numbers in {curly braces} are from the magnificent Slice of Life videodisk. No medical student should be without access to this wonderful resource. Someday you may be able to access these pictures directly from this page.
Also:
Medmark Pathology -- massive listing of pathology sites
Freely have you received, freely give. -- Matthew 10:8. My
site receives an enormous amount of traffic, and I'm
handling about 200 requests for information weekly, all
as a public service.
Pathology's modern founder,
Rudolf
Virchow M.D., left a legacy
of realism and social conscience for the discipline. I am
a mainstream Christian, a man of science, and a proponent of
common sense and common kindness. I am an outspoken enemy
of all the make-believe and bunk that interfere with
peoples' health, reasonable freedom, and happiness. I
talk and write straight, and without apology.
Throughout these notes, I am speaking only
for myself, and not for any employer, organization,
or associate.
Special thanks to my friend and colleague,
Charles Wheeler M.D.,
pathologist and former Kansas City mayor. Thanks also
to the real Patch
Adams M.D., who wrote me encouragement when we were both
beginning our unusual medical careers.
If you're a private individual who's
enjoyed this site, and want to say, "Thank you, Ed!", then
what I'd like best is a contribution to the Episcopalian home for
abandoned, neglected, and abused kids in Nevada:
My home page
Especially if you're looking for
information on a disease with a name
that you know, here are a couple of
great places for you to go right now
and use Medline, which will
allow you to find every relevant
current scientific publication.
You owe it to yourself to learn to
use this invaluable internet resource.
Not only will you find some information
immediately, but you'll have references
to journal articles that you can obtain
by interlibrary loan, plus the names of
the world's foremost experts and their
institutions.
Alternative (complementary) medicine has made real progress since my
generally-unfavorable 1983 review linked below. If you are
interested in complementary medicine, then I would urge you
to visit my new
Alternative Medicine page.
If you are looking for something on complementary
medicine, please go first to
the American
Association of Naturopathic Physicians.
And for your enjoyment... here are some of my old pathology
exams
for medical school undergraduates.
I cannot examine every claim that my correspondents
share with me. Sometimes the independent thinkers
prove to be correct, and paradigms shift as a result.
You also know that extraordinary claims require
extraordinary evidence. When a discovery proves to
square with the observable world, scientists make
reputations by confirming it, and corporations
are soon making profits from it. When a
decades-old claim by a "persecuted genius"
finds no acceptance from mainstream science,
it probably failed some basic experimental tests designed
to eliminate self-deception. If you ask me about
something like this, I will simply invite you to
do some tests yourself, perhaps as a high-school
science project. Who knows? Perhaps
it'll be you who makes the next great discovery!
Our world is full of people who have found peace, fulfillment, and friendship
by suspending their own reasoning and
simply accepting a single authority that seems wise and good.
I've learned that they leave the movements when, and only when, they
discover they have been maliciously deceived.
In the meantime, nothing that I can say or do will
convince such people that I am a decent human being. I no longer
answer my crank mail.
This site is my hobby, and I presently have no sponsor.
This page was last updated February 6, 2006.
During the ten years my site has been online, it's proved to be
one of the most popular of all internet sites for undergraduate
physician and allied-health education. It is so well-known
that I'm not worried about borrowers.
I never refuse requests from colleagues for permission to
adapt or duplicate it for their own courses... and many do.
So, fellow-teachers,
help yourselves. Don't sell it for a profit, don't use it for a bad purpose,
and at some time in your course, mention me as author and KCUMB as my institution. Drop me a note about
your successes. And special
thanks to everyone who's helped and encouraged me, and especially the
people at KCUMB
for making it possible, and my teaching assistants over the years.
Whatever you're looking for on the web, I hope you find it,
here or elsewhere. Health and friendship!
If you were hatched from a swan's egg, it doesn't matter that you may have begun life in a chicken coop.
-- Hans Christian Anderson
When you see a person who has been given more than you in money or beauty, then look to those who
have been given less.
-- Mohammed
We [the human race] do not have much time to prove that we are not the product of a lethal mutation.
-- Science 263: 181, 1994
 I am presently adding clickable links to
images in these notes. Let me know about good online
sources in addition to these:
I am presently adding clickable links to
images in these notes. Let me know about good online
sources in addition to these:
Pathology Education Instructional Resource -- U. of Alabama; includes a digital library
Houston Pathology -- loads of great pictures for student doctors
Pathopic -- Swiss site; great resource for the truly hard-core
Syracuse -- pathology cases
Walter Reed -- surgical cases
Alabama's Interactive Pathology Lab
"Companion to Big Robbins" -- very little here yet
Alberta
Pathology Images --hard-core!
Cornell
Image Collection -- great site
Bristol Biomedical
Image Archive
EMBBS Clinical
Photo Library
Chilean Image Bank -- General Pathology -- en Español
Chilean Image Bank -- Systemic Pathology -- en Español
Connecticut
Virtual Pathology Museum
Australian
Interactive Pathology Museum
Semmelweis U.,
Budapest -- enormous pathology photo collection
Iowa Skin
Pathology
Loyola
Dermatology
History of Medicine -- National Library of Medicine
KU
Pathology Home
Page -- friends of mine
The Medical Algorithms Project -- not so much pathology, but worth a visit
National Museum of Health & Medicine -- Armed Forces Institute of Pathology
Telmeds -- brilliant site by the medical students of Panama (Spanish language)
U of
Iowa Dermatology Images
U Wash
Cytogenetics Image Gallery
Urbana
Atlas of Pathology -- great site
Visible
Human Project at NLM
WebPath:
Internet Pathology
Laboratory -- great site My team:
My team:Ed Lulo's Pathology Gallery
Bryan Lee's Pathology Museum
Dino Laporte: Pathology Museum
Tom Demark: Pathology Museum
Dan Hammoudi's Site
Claude Roofian's Site
Pathology Handout -- Korean student-generated site; I am pleased to permit their use of my cartoons
Estimating the Time of Death -- computer program right on a webpage
Pathology Field Guide -- recognizing anatomic lesions, no pictures

St.
Jude's Ranch for Children
I've spent time there and they are good. Write "Thanks
Ed" on your check.
PO Box 60100
Boulder City, NV 89006--0100
More of my notes
My medical students
Clinical
Queries -- PubMed from the National Institutes of Health.
Take your questions here first.
HealthWorld
Yahoo! Medline lists other sites that may work well for you

We comply with the
HONcode standard for health trust worthy
information:
verify
here. 

![]()
Inherited genetic disease is of tremendous importance in clinical medicine. Except as marked or noted in lecture, this is mastery-level material at the recall level.
You should be able to:
QUIZBANK
Developmental abnormalities (all);
Metabolic #'s 5-8, 25-41, 117
 KEY IDEAS AND TERMS
KEY IDEAS AND TERMS
* "An organism is the genes' way of making more genes." Today, no life-scientist seriously doubts that the tremendous diversity and success of living creatures is due, at least in part, to natural selection for and against genes that have been randomly altered. Since most mutations probably confer no advantage or disadvantage (Nature 354: 114, 1991), random drift ("the molecular clock") also plays an important role in changing the sequences.
* A species is a breeding population that can produce fertile offspring among its members, but not outside its group. We know that new species arise as a result of reproductive isolation; members of the ancestral populations have become separated by geography and/or niche. The molecular mechanisms that underlie micro-evolution (i.e., loss of the ability to interbreed) must be the result of genetic drift in an isolated population, so that at least one enzyme no longer meshes with its distant cousins. We look forward to learning the details, but obviously this is a long way off.
The ability to evolve, and to select helpful genes, is basic to the survival of life on earth. Humankind pays a price for the tendency of the genes to keep changing. The cost is borne by the "less fit", especially the 7% (estimates vary, and this doesn't include "polygenic inheritance") of people with significant genetic disease, and by those who care for them. Most embryonic and early fetal wastage is probably due to genetic and chromosomal problems, and even today, we don't know how common these losses really are.
 You know about genes, alleles, chromosomes (autosomal and sex), mitosis and meiosis,
haploid and
diploid cells (exact multiples of the haploid number are euploid; others are aneuploid), mutations
(and
the environmental problems that cause them), centromeres, and the basic biology of nucleic acids. You
also understand classic Mendelian and sex-linked inheritance, homozygosity, heterozygosity,
hemizygosity, and consanguineous mating. If any of these terms are unfamiliar, please review. If you
don't know what restriction fragment length polymorphism is all about, ask a molecular diagnostician --
it's important.
You know about genes, alleles, chromosomes (autosomal and sex), mitosis and meiosis,
haploid and
diploid cells (exact multiples of the haploid number are euploid; others are aneuploid), mutations
(and
the environmental problems that cause them), centromeres, and the basic biology of nucleic acids. You
also understand classic Mendelian and sex-linked inheritance, homozygosity, heterozygosity,
hemizygosity, and consanguineous mating. If any of these terms are unfamiliar, please review. If you
don't know what restriction fragment length polymorphism is all about, ask a molecular diagnostician --
it's important.
Remember that germ line mutations are present in the sperm or the egg, while somatic mutations are acquired after fertilization.
Notice that if two normal parents give birth to a child with achondroplasia, one of them either had a somatic mutation involving the germinal epithelium, or a mutation involving just a single gamete.
Certain diseases of the genes can result only from post-zygotic, somatic mutations. These include McCune-Albright's syndrome (rare) and the fully-expressed malignant phenotype (all too common).
| Hutchinson-Gilford progeria ("accelerated aging") is apparently always caused by a de novo mutation in a sperm (Science 300: 1995, 2003). More about this later. |
|
Vertical transmission of a mutation occurs from parent to child. Horizontal transmission of a mutation occurs within a single organism as a clone of mutated cells (as in tumors, and such mosaics as McCune-Albright's). As we will soon see, diseases of DNA are really of two types -- inherited disease (vertical transmission) and tumors (horizontal transmission). Nowadays, we call only the first "genetic disease".
The genetic code translates nucleic acid base pair triplets (codons) into animo acids for a protein sequence. It is almost (not exactly: Comp. Bio. Phys. 106: 89, 1993; J. Mol. Evo. 34: 331, 1992) constant across nature, and entirely constant for the nuclei of the higher creatures. (* This is one more piece of evidence that evolution has occurred on the grand scale, since the code is apparently arbitrary.)
Restriction enzymes (endonucleases): Enzymes, mostly from bacteria, that cleave ("digest") DNA at specific sequences. * EcoR1, still the "champ", cleaves at GAATTC.
Denatured DNA: single-stranded DNA
Exon: the portion of the gene that is transcribed onto mRNA, for translation into protein sequences.
Intron: the rest of the DNA in the gene under discussion.
Gene probe: a single-stranded nucleic acid sequence, labeled with radio-isotope, used to identify a specific complementary sequence
Southern blotting: using probes to search for particular DNA sequences, on digested DNA samples that have been digested using restriction enzymes and the fragments separated by electrophoresis. Invented by Dr. Southern.
Northern blotting: searching for m-RNA by a technique similar to Southern blotting. Named by some wag in imitation of "Southern".
Western blotting: searching for a protein, using electrophoretic methods
Dot blotting: like Northern and Southern blotting, only without using electrophoresis
In-situ hybridization: using probes to detect nucleic acid sequences within cells, without destroying the cells themselves.
Polymerase chain reaction: a technique to identify very small quantities (perhaps even a single copy) of a particular DNA sequence in a sample. This has many uses, ranging from the most sensitive and specific AIDS test to a way of telling whether a leukemia is completely cured (Lancet 344: 348, 1994).
Classical genetic research: define the biochemical abnormality, then isolate the protein, sequence it, and identify the gene. How we cloned the hemoglobin S gene. "Functional cloning."
Reverse genetic research (positional cloning): locate the affected chromosome, sequence until you find the gene, then deduce the protein sequence ("the predicted protein") and find it, and finally figure out its function. Duchenne's muscular dystrophy, cystic fibrosis, and Li-Fraumeni disease were all successfully approached in this way. "Positional cloning."
Pseudogene: a DNA sequence, once useful (we may suppose) or else transferred from a real gene, and homologous to active genes in us or in related organisms, but now genetically inactive. Pseudogenes left over from evolution would constitute the ultimate "vestigial structures".
Genetic disease: almost impossible to define. My best shot is, "a disease that is determined, more or less, the moment the egg is fertilized." ("You made the first mistake, you picked the wrong parents.")
Entities such as sickle cell disease and Huntington's chorea develop when, and only when, a particular gene is defective. Yet even sickle cell disease is modified by the presence or absence of thalassemia genes. Cystic fibrosis, a simple mendelian problem, varies in severity depending on which of more than a dozen alleles has been inherited.
Identical twins are 100% concordant for type II diabetes, which is polygenic, and the course of the disease is influenced by environmental factors.
Diseases like lupus, high blood pressure, alcoholism, and schizophrenia show obvious familial tendencies, even in siblings raised apart, yet many identical twins are spared.
The variability in particular diseases from person to person reflects, in part, our varying genetic heritage.
A handful of diseases, outstandingly McCune-Albright syndrome, Klippel-Trenaunay, Ollier/Maffucci, and Proteus syndrome, are genetic diseases that cannot be inherited, but that always result from a mutation early in embryogenesis.
Even the "Big Robbins" example of automobile accidents as a process in which "the environment totally determines the nature of disease" ignores the obvious genetic factors in alcoholism. (Rabies or gonorrhea would be better examples.)
Notice that the above definitions of "genetic disease" ignore the other, equally important, "acquired genetic diseases", i.e., tumors, in which defective genes are propagated and accumulate within clones of cells in a single organism. We now talk about inherited genetic disease ("vertical transmission") and neoplastic genetic disease ("lateral transmission"). Much more about this soon.
Hereditary disease: "Genetic disease", with the exception of those rare ones you can't inherit.
Familial disease: Definitions vary. "Diseases that cluster within families" can include classic genetic diseases, polygenic disease, mysterious things (more on "SIDS" later in the course), heritable viruses, nutritional stuff, environmental stuff ("Is that lead paint peeling from the walls?"), and, of course, behavioral stuff -- the child-abuse cycle, obesity ("Clean your plate!"), etc., etc.
Congenital disease: a disease present at birth. Note that sickle cell disease and Huntington's chorea, both genetic, are not symptomatic at birth, and that various traumas and infections acquired in utero are congenital but not genetic.
Transgenic mice: mice developed from fertilized eggs in which the genetic material was manipulated.
Knockout mice have had genes deleted, which is tricky.
Chromosomal aberration ("cytogenetic aberration"): diseases in which there are the wrong number of copies of some or all of a chromosome, sufficient to detect using classic karyotyping. About 1% of newborns, and maybe 50% of spontaneous abortions, possess an abnormal karyotype.
G-banding (from "Giemsa" stain): a technique that visualizes bands on chromosomes, improving our ability to localize genes and recognize minor karyotypic problems.
Standard cytogenetic terminology names a karyotype for the number of chromosomes, a list of the sex chromosomes, and mention of any extra ("+") or deleted ("-") chromosomes. Abnormalities of individual chromosomes are designated by "p" for the short arm, "q" for the long arm, and mention of the numbered regions shown by G-banding.
Imprinting: Genes and chromosomes sometimes differ slightly depending on whether they were acquired from Mom or Dad. A hot topic right now.
Uniparental disomy: Non-mendelian inheritance in which two copies of a gene or chromosome were inherited from the same parent.
Triplet (trinucleotide) repeat mutations: Genes rendered abnormal by the amplification of CG-rich units. This is the usual mechanism of mutation in fragile-X disease, Huntington's, and myotonic dystrophy, and a few others. Unlike more familiar syndromes, these diseases get worse from generation to generation as the amplification continues (anticipation or Sherman's paradox, long-noted in myotonic dystrophy, means the disease appears sooner in the son than in the father).
Mutant genes of large effect: diseases caused by a single defective gene.
Most are Mendelian disorders, either autosomal or sex-linked. We are now discovering diseases caused by mutated mitochondrial genes.
* It is simply wrong to think of all mutations as just rendering a gene ineffective. The old pseudoscientist's complaint, "How can a mutation give rise to something useful?" is now abundantly invalidated by studies of the origins of modern genes from mutations (base-pair substitutions, recombinations, even frame-shifts; see Nature 306: 203, 1983) of genes that once did something else.
Sex-limited inheritance: diseases inherited independent of the sex-chromosomes, but that can ordinarily express themselves only in one sex or the other. The prime example is male baldness -- a woman must usually take exogenous testosterone in order to go bald.
Polygenic inheritance: diseases that are caused or significant modulated by several different abnormal genes
Multifactorial etiology: despite "Big Robbins", this may mean either "caused by several abnormal genes" or "requiring both abnormal genes and an abnormal environment".
Lyonization: Inactivation, early in embryogenesis, of all but one of the X-chromosomes in each cell. Once lyonization has occurred, the same X-chromosome will be inactivated in all of that cell's progeny, where it will be the Barr body (or bodies, for those with more than two X's), or sex chromatin (visible on buccal smear), until oogenesis is required again.
|
{13429} Barr body (11 o'clock) |
Note that the entire chromosome is not inactivated. (If the second X were totally inactive, then XO's, XXY's, and XXXX's would be phenotypically normal. * The Kallmann's syndrome gene (no olfactory nerves, no gonadotropins, all because of defective neuronal migration, see NEJM 326: 1752, 1992), located on X (with an inactive counterpart on Y), is expressed whether or not the chromosome is lyonized (Nature 383: 529, 1991).
Lyonization may be lucky or unlucky. For example, female identical twins with one X-chromosome carrying color blindness may be discordant for color blindness (Am. J. Hum. Genet. 51: 291, 1992).
If the X's are discordant for skin color (for whatever reason), you will end up spotted (Arch. Derm. 129: 1460, 1993). Whenever X's differ in some significant way, the woman is a "functional mosaic" (in contrast to a "genomic mosaic", caused by a somatic mutation).
* Transcription of a gene from the inactivated X: Nature 351: 325, 1991. By the way, we're still puzzled about the actual molecular biology of lyonization itself. The gene XIST produces an RNA that ties up the lyonized chromosome, and methylation of cytosines also seems to be involved (Nature 368: 154, 1994, others).
Pseudo-autosomal inheritance: The tips of the short arms of chromosomes X and Y are homologous, and a few genes (* notably blood group Xg) are located here (Am. J. Hum. Genet. 51: 1172, 1992).
Somatic mosaicism: when all cells in a person do not have essentially the same genetic makeup. This can result from a mutation in one cell during the early stages of embryogenesis, or even from fusion of two fertilized eggs to produce one person (i.e., a chimera -- * ponder that!, fortunately, it's rare.)
Chimerism may also result from blood exchange between fraternal twins, and the person has two people's bone marrow. Future blood bankers: these patients will probably have two different blood types.
Somatic mosaicism probably underlies many (if not most) birthmarks (for example Lancet 345: 596, 1995). People living comfortably with a "forme fruste" of some dreadful classic lethal disease may be mosaics (for example, Am. J. Hum. Genet. 46: 591, 1990). It's also the basis of tumorigenesis (more about this later).
Functional mosaicism is the result of lyonization, as explained above. All about mosaics: Arch. Derm. 129: 1460, 1993 (why a dermatologist?)
Germinal mosaicism means that a mother who is not affected by a particular mutation has transmitted it more than once as if she were; i.e., the eggs arose from a mutant clone.
Penetrance: the chance that someone with the gene (or genes) for a condition will express the condition.
Variable expressivity: a term for an allele that causes widely different degrees of abnormality in different people.
Balanced polymorphism: the heterozygote enjoys an advantage that has allowed selection for the gene, making the homozygous condition common. The prime example is sickle cell disease, in which the heterozygote enjoys immunity to malaria. Cystic fibrosis, Gaucher's, and probably others are probably also balanced polymorphisms.
Pleiotropism: one mutant gene produces several effects
Genetic heterogeneity: the same effect can be produced by mutations at several sites.
For example, there are several different loci at which two defective genes will produce an albino, there are at least two genes that produce adult polycystic kidney disease, there are at least seven ways to get xeroderma pigmentosum, there are multiple dominant and recessive forms of retinitis pigmentosa (Am. J. Hum. Genet. 53: 80, 1993; Nat. Genet. 23: 217, 1999, lots more), there are about a dozen known ways to get Ehlers-Danlos syndrome, there are several dozen deafness syndromes, etc., etc.
* My favorite example, right now, is Leigh's syndrome, a progressive brain disease of young children in which there is defective cytochrome oxidase in the mitochondria. Sounds straightforward -- except that the cytochrome oxidase complex is coded by at least 13 different genes, some in the nucleus and some in the mitochondria. (For starters, three different ways to get Leigh's are described in Ped. Res. 26: 260, 1989, J. Ped. 116: 84, 1990, and Neurology 39: 697, 1989). Probably there are many, many alleles here, and many Leigh's cases could even be polygenic.
Dermatoglyphics: examining the lines on the palms and soles, and the fingerprint ridge patterns. A fascinating game that has yielded many interesting correlations, none of any clinical utility. For a nice review, see J. Invest. Dermatol. 43: 261, 1970. (* P.S. As a pathology resident, I used to check the lengths of the "life lines" on autopsy patients' palms, to see how long they had lived. Sorry, no obvious correlation.)
* Genetic load: the frequency of deleterious recessive mutations in a population. An important concept in biology. In populations with high genetic load (i.e., most humans), matings of near relatives is likely to result in defective newborns. By contrast, in populations with low genetic load (i.e., lab animals, many wild populations), inbreeding is not a hazard. Frequent inbreeding does reduce genetic load in the long run. Remember that Cleopatra ("the most beautiful woman in history", they say) was the offspring of several sequential brother-sister matings.
* In my mind, God wrote two books. The first book is the Bible, where humans can find the answers to their questions on values and morals. The second book of God is the book of nature, which allows humans to use observation and experiment to answer our own questions about the universe.
-- Galileo, to the Grand Duchess of Tuscany
* Thus, from the war of nature, from famine and death, the most exalted object which we are capable of conceiving, namely the production of the higher animals, directly follows. There is a grandeur in this view of life, with its several powers, having been originally breathed by the Creator into a few forms or into one; and that, whilst this planet has gone cycling on according to the fixed law of gravity, from so simple a beginning endless forms most beautiful and most wonderful have been, and are being evolved.
-- Charles Darwin, The Origin of Species, 1860, emphasis added
* Today, more than a half century after [Humani Generis], new knowledge leads us to recognize in the theory of evolution more than a hypothesis. ... The convergence, neither sought nor induced, of results of work done independently one from the other, constitutes in itself a significant argument in favor of this theory. ... The elaboration of a theory such as that of evolution, while obeying the exigency of homogeneity with the data of observation, borrows certain ideas from the philosophy of nature. ... There are thus materialistic and reductionist readings and spiritual readings. ... Pius XII underlined this essential point: If the origin of the human body is sought in living matter which existed before it, the spiritaul soul is directly created by God."
-- Pope John Paul II
* The serious questions most often asked of me by students concern the history of life. Before I placed this short, non-testable section, I would be questioned many times each year. You asked, so here goes.
* By today's standards, I'd be considered an old-earth, soft creationist with a high regard for truth, and who demands basic honesty and ordinary decency from others. My longstanding interest in the philosophic questions raised, and my contribution, are known, and this is rumor control.
* Trying to do biology without talking about common descent is like trying to do chemistry without talking about the periodic table. Yet the language makes for misunderstandings. When a scientist says "evolution", he/she simply means "common descent". When religionists (or militant anti-religionists) use the term "evolution", they usually mean "scientific materialism" or "naturalistic reductionism" or "naive naturalism", i.e., the idea that the success of physics, chemistry, and biology in explaining and predicting things that happen in the material world rules out the existence of the supernatural.
* And you already know that when a scientist says "true" or "I believe", he/she is simply expressing confidence in the power of a claim to predict future observations.
* You don't have to "believe in evolution" (or even common descent) if you don't want to. I would prefer to believe a literal reading of Genesis. But there are things that you can observe in our world that I don't think you can explain unless we share a common ancestry with other living things, and the theory's predictive power speaks for itself. During the creation science battles of the early 1980's, I catalogued 38 distinct positions on "the origins question" taken by serious Christians, past and present. I decided that there were problems with each of these, but that they might not all be unsurmountable.
* In the meantime, if you have ever had pets, you know how much they have in common with us. They possess many qualities that we admire, often being good in ways that few humans are. It puzzles (and upsets) me that some religionists object to the idea that we are descended from animals. Perhaps Christian belief is actually reflected best in Christian art. Only humans sin, so only humans need the Cross. The newly-incarnate Christ is usually shown surrounded by animals, and better Christians than myself have argued that they are saved by His Incarnation.
* (1) Most of the interesting work in evolution ("inference of phylogenetic relationships") is now done in molecular biology rather than in paleontology (for example, bird-lovers will enjoy Proc. Nat. Acad. Sci. 91: 2621, 1994; those who prefer bats will want to read Science 256: 86, 1992; origin of dogs (they go way back, maybe 100,000 years Science 276: 1647, 1997); Afrotheria (elephants, sea cows, hyraxes evolved together in Africa when it was isolated; Proc. Nat. Acad. Sci. 98: 2001; how we diverged Proc. Nat. Acad. Sci. 100: 1062, 2003); for those who like people best, see Nat. Genet. 33-S: 266, 2003. common origin of immunoglobulins and T-cell receptors Proc. Nat. Acad. Sci. 93: 3289, 1996; reconstructing the history of lysozyme Nature 385: 151, 1997; origin of eukaryotes Science 257: 30 & 74, 1992; and all the way back, using tRNA synthetase Proc. Nat. Acad. Sci. 92: 2441, 1995). At our present state of knowledge, much of our nucleic-acid genome appears to be "junk", left over from our past, or even of viral origin -- the ultimate successful parasitization. If we truly share ancestry with chimpanzees, we would predict that our true junk DNA sequences are homologous, almost (but not quite) as strikingly similar as the two species' known genes and chromosomes. And it is. For example, the non-coding section of one early-studied chimp gene is a 98.4% match to its human counterpart, much closer to the human than to the gorilla (Science 250: 376, 1990; chimp update Science 302: 1960, 2003). In fact, if you look at our DNA, we're as similar to the two species of chimps as they are to each other (Science 263: 181, 1994). Enough is enough. I'm satisfied this is common ancestry, not just "common design".
* (2) You all know the genetic code is degenerate. The study of "silent sites" (redundant third base pairs of codon triplets) is getting to be a discipline all by itself. These shouldn't make the slightest difference as far as the creature is concerned (see J. Mol. Evo. 33: 442, 1991; J. Mol. Evo. 36: 201, 1993), yet the closer-related the animals are by classic evolutionary study, the closer the "silent sites" match, just as Darwin's theory predicts (and if it were not true, Darwin's theory would be refuted); hard to explain if nucleic acid sequences were created separately for the garden of Eden. See Proc. Nat. Acad. Sci. 88: 5974, 1991; Mol. Bio. Evo. 9: 193, 1992 (insulin); J. Mol. Evo. 33: 442, 1991; J. Mol. Evo. 37: 441, 1993 (rats and mice differ in silent site G+C by only 1.7%, much less than even their amino acid sequences). Line up any comparison of the base sequence for the same protein in two different creatures (go search the literature yourselves), and you'll find this to be true.
 * (3) Most people
do not understand science,
and this is not their fault.
Most religionists are good, decent people.
I note with much
satisfaction that classic creationism (i.e., the versions that deny common
descent)
is now confined to ultra-Right wing circles
and rare New Agers. During the trials that culminated in "Aguillard",
all of the major "creation scientists"
consistently refused to testify under oath,
and despite their soliciting funds for "research" and complaining that they
cannot get a fair hearing, not one of them was able to produce a single
letter of rejection from a refereed journal. In other words, it was (and still is)
all fake.
Please be careful, especially if you're asked
for money. There are still some utterly cynical
people operating.
In a country like ours, in which most adults can't tell you why the
seasons occur, pseudoscience (tricky fallacies, falsified evidence, "we're spiritual and the working
scientists are immoral and anti-God") makes easy bucks.
All of the following are lies:
"Evolution is not true because transitions in the fossil record are abrupt."
"Darwin renounced evolution on his deathbed." "There aren't enough known human
fossils to cover a pool table." "A modern human skull was found in dinosaur strata [the original report
was from an old US tabloid newspaper]." "Scientists [actually, just the staff of another old London
tabloid newspaper, which
I have] mistook a pig tooth for Nebraska Man." "For decades, the Piltdown fraud
was central to thinking about human origins."
(Check out an old textbook -- it was considered an unexplained anomaly
before the fraud was revealed.)
"Human Cytochrome C is closer to a chicken (Answers in Genesis) or a
sunflower (Kent Hovind) than to a chimp."
"Human cholesterol is more like that of lower life forms than it's like an ape's."
"There's only 6000 years worth of dust on the moon's surface." "At Paluxy, Texas, human footprints
were found in dinosaur strata" [1930's creationists carved these themselves as a deliberate, confessed
hoax]. "Believing in evolution is the primary cause of racism and all other contemporary evils."
"Evolution violates the second law of thermodynamics." "If you simply drop the assumption that the
speed of light is constant over time, then the universe could be only 6000 years old."
"Red shift and the cosmic background radiation could be explained by tired
light." "The NASA
computer discovered a day was missing from the past and this was because Joshua made the sun stand
still." "If scientists really believed we're more closely related to chimps
than rats, they wouldn't use rats for research." "Transitional forms should appear at a constant rate."
"There is a massive evil conspiracy to...." And so on, ad nauseam.
In racist South Africa in the 1940's-1980's, the
lies were even goofier and uglier.
The most popular young-earth creationist right now also sells laetrile,
the phony cancer medicine from the 1970's, and "The Protocols of the Learned
Elders of Zion", a rabid anti-semitic forgery from the Nazi era.
If you want to be involved with people like this, that is your business.
I judge a person's sincerity by his or her willingness to testify
in a court of law, under oath. These operators are notoriously
unwilling to do so.
At the Arkansas trial,
the only creationist witness on genetics was Chandra Wickramasinghe, a Ceylonese-Welsh
astrophysicist. Professor Wickramasinghe testified that he believed insects were more intelligent than
humans, so much so that they were keeping it a secret. I am not making any of this up.
Geneticist
Francis "Discoverer of the Cystic Fibrosis Gene Etc. and Long-Time
Head of the NIH Human Genome Research Institute" Collins, a conservative Evangelical, noted in Physician
6(3), 1996 ("Focus on the Family", a conservative-Christian publication) that his fellow-scientists are
simply wrong to assume that, because he's a Christian, he's also a
classical creationist. Of course he's not.
* (3) Most people
do not understand science,
and this is not their fault.
Most religionists are good, decent people.
I note with much
satisfaction that classic creationism (i.e., the versions that deny common
descent)
is now confined to ultra-Right wing circles
and rare New Agers. During the trials that culminated in "Aguillard",
all of the major "creation scientists"
consistently refused to testify under oath,
and despite their soliciting funds for "research" and complaining that they
cannot get a fair hearing, not one of them was able to produce a single
letter of rejection from a refereed journal. In other words, it was (and still is)
all fake.
Please be careful, especially if you're asked
for money. There are still some utterly cynical
people operating.
In a country like ours, in which most adults can't tell you why the
seasons occur, pseudoscience (tricky fallacies, falsified evidence, "we're spiritual and the working
scientists are immoral and anti-God") makes easy bucks.
All of the following are lies:
"Evolution is not true because transitions in the fossil record are abrupt."
"Darwin renounced evolution on his deathbed." "There aren't enough known human
fossils to cover a pool table." "A modern human skull was found in dinosaur strata [the original report
was from an old US tabloid newspaper]." "Scientists [actually, just the staff of another old London
tabloid newspaper, which
I have] mistook a pig tooth for Nebraska Man." "For decades, the Piltdown fraud
was central to thinking about human origins."
(Check out an old textbook -- it was considered an unexplained anomaly
before the fraud was revealed.)
"Human Cytochrome C is closer to a chicken (Answers in Genesis) or a
sunflower (Kent Hovind) than to a chimp."
"Human cholesterol is more like that of lower life forms than it's like an ape's."
"There's only 6000 years worth of dust on the moon's surface." "At Paluxy, Texas, human footprints
were found in dinosaur strata" [1930's creationists carved these themselves as a deliberate, confessed
hoax]. "Believing in evolution is the primary cause of racism and all other contemporary evils."
"Evolution violates the second law of thermodynamics." "If you simply drop the assumption that the
speed of light is constant over time, then the universe could be only 6000 years old."
"Red shift and the cosmic background radiation could be explained by tired
light." "The NASA
computer discovered a day was missing from the past and this was because Joshua made the sun stand
still." "If scientists really believed we're more closely related to chimps
than rats, they wouldn't use rats for research." "Transitional forms should appear at a constant rate."
"There is a massive evil conspiracy to...." And so on, ad nauseam.
In racist South Africa in the 1940's-1980's, the
lies were even goofier and uglier.
The most popular young-earth creationist right now also sells laetrile,
the phony cancer medicine from the 1970's, and "The Protocols of the Learned
Elders of Zion", a rabid anti-semitic forgery from the Nazi era.
If you want to be involved with people like this, that is your business.
I judge a person's sincerity by his or her willingness to testify
in a court of law, under oath. These operators are notoriously
unwilling to do so.
At the Arkansas trial,
the only creationist witness on genetics was Chandra Wickramasinghe, a Ceylonese-Welsh
astrophysicist. Professor Wickramasinghe testified that he believed insects were more intelligent than
humans, so much so that they were keeping it a secret. I am not making any of this up.
Geneticist
Francis "Discoverer of the Cystic Fibrosis Gene Etc. and Long-Time
Head of the NIH Human Genome Research Institute" Collins, a conservative Evangelical, noted in Physician
6(3), 1996 ("Focus on the Family", a conservative-Christian publication) that his fellow-scientists are
simply wrong to assume that, because he's a Christian, he's also a
classical creationist. Of course he's not.
I see no conflict in what the Bible tells me about God and what science tells me about nature. Like St. Augustine in AD 400, I do not find the wording of Genesis 1 and 2 to suggest a scientific textbook but a powerful and poetic description of God's intention in creating the universe. The mechanism of creation is left unspecified. If God, who is all powerful and who is not limited by space and time, chose to use the mechanism of evolution to create you and me, who are we to say that wasn't an absolutely elegant plan? And if God has now given us the intelligence and the opportunity to discover hismethods, that is something to celebrate.I lead the Human Genome Project, which has now revealed all of the 3 billion letters of our own DNA instruction book. I am also a Christian. For me scientific discovery is also an occasion for worship.
Nearly all working biologists accept that the principles of variation and natural selection explain how multiple species evolved from a common ancestor over ery long periods of time. I find no compelling examples that this process is insufficient to explain the rich variety of life forms present on this planet. While no one could claim yet to have ferreted out every detail of how evolution works, I do not see any significant "gaps" in the progressive development of life's complex structures that would require divine intervention. In any case, efforts to insert God into the gaps of contemporary human understanding of nature have not fared well in the past, and we should be careful not to do that now.
Science's tools will never prove or disprove God's existence. For me the fundamental answers about the meaning of life come not from science but from a consideration of the origins of our uniquely human sense of right and wrong, and from the historical record of Christ's life on Earth.
-- Francis Collins "Time" August 2005
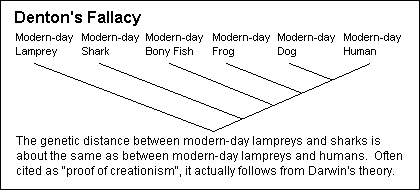
I will abandon my belief in common descent if a single protein family is found that grossly violates the Darwinist prediction that its phylogenetic history revealed in its base-pair sequences, notably its choice of degenerate codons, will match the history of life suggested by classic comparative anatomy. If they believed their own claims, they would be sequencing genes. My best "thought question" for an acceptable medical school candidate is to explain Denton's Fallacy ("Evolution: A Theory in Crisis"): Why is the genetic distance between a modern-day shark and a modern-day bony fish the same as the genetic distance between a modern-day shark and a human being? The most-cited post-Aguillard vs. Edwards "evidence for creation", it actually follows from Darwin's theory. How? Behe's Fallacy is a restatement at the molecular level of the old claim that bodyparts are interdependent and hence could not have arisen individually without "intelligent design". The answer is that they evolve together. Behe's most familiar claim is that the clotting cascade is "irreducibly complex" and with today's knowledge of the clotting cascades of other animals, this is a lie, pure and simple. Behe was torn to pieces on cross-examination during the 2005 trial. (The other "intelligent design" proponents were smart enough to refuse to testify.) Every last Nobel science laureate in the U.S. signed an amicus brief to the Supreme Court in "Aguillard vs. Edwards", testifying that classic "creation science" is a mass of untruths by sectarian ideologues seeking political power. This is the only time they have all agreed about anything. Even before Aguillard, a friend of mine went to buy some creationist books at what he told me was the largest Christian bookstore in northern California. They didn't stock any, and the owner said, "We don't carry that cr_p." If you must deny common descent, you're best to stick to the "omphalos" (Adam was created as an adult with a belly-button) argument, i.e., "God created the fossils and the other evidence for evolution to deceive people so they would go to hell." Still popular in some "conservative" circles; I'm glad I don't believe it.
* (4) In striking contrast to the classic creationists, today's thinking Evangelicals seem to have come up with a new synthesis that fits the scientific facts so far. Especially if you think that "believing in evolution" will be bad for your spiritual life, or make you immoral or whatever... let me recommend progressive creationism instead of the more classic versions. "Progressive creationism" is several positions, all long-known in Christian circles, and anathematized by the better-known (i.e., big-money) creationists; the most popular version today is that God directed the mutations and/or added genes in the gametes of animals to lead to humankind. (To my knowledge, no progressive creationist has advanced the idea that God worked primarily by protecting individuals with random mutations that were intended to lead ultimately to humans. I guess somebody might say "Hey, that sounds like natural selection!") Progressive creationism fits the available data, and I also recommend it if you're impressed by "design in nature..." or "the odds against..." or "we still can't explain the origins of the genetic code..." (I am not impressed, preferring the parsimony of the Modern Synthesis, but I have no wish to argue with you over something so subjective). "Progressive creationism" (in the version I've just described) is now mainstream throughout most of conservative Christendom. It is also usual for these people to claim that life could not have arisen by natural processes. (Dr. Behe's deceitfully-entitled book "Darwin's Black Box" misidentifies Darwin's theory and the idea that the genetic code itself has a natural origin.) At Christian bookstores, you can buy high-quality books about geology and biology that could pass for secular science books (the earth is 4.5 billion years old, plate tectonics and all, common descent is quietly acknowledged), except for a few paragraphs about how "life could not have arisen by chance." At present (2004) the two most popular creationist ("intelligent design" nowadays) writers (Lee Strobel and Philip Johnson) are both attorneys, not zoologists or paleontologists or geneticists. Attorneys are specially educated to create confusion and misrepresent what others have said in order to make people believe things that even the attorney may know are not true. The two other major writers are William Dembski, mathematician and Jonathan Wells, who is the one writer who has been promoted as a bona-fide bioscientist. Wells is actually one of twelve seminarians from the the Unification Church who, at the request of Sun Myung Moon, were sent through a graduate program in biology in order to promote creationism once they were finished (Nature 410: 745, 2001). Wells doesn't mention this in his biographical sketch. (Morals, guys?) Anyone who knows basic biology can see though these guy's' fallacies. And all of them are presenting progressive creationism, not the familiar "Noah's ark" stuff. If you read the "intelligent design" stuff, you will notice that the authors never address the question of humans have a non-human ancestor. By now, one of the attorneys has become a laughing-stock for his evasiveness when asked directly whether human beings have non-human ancestors, whether the first mouse had a mother, and so forth. (See Martin Gardner's review in the Skeptical Inquirer, 1998). And even the influential Charles Colson chose to quote another attorney rather than a scientist in his book on answering kids' questions, telling them why they should not believe in evolution. If progressive creationism is true, then it could easily be confirmed (and God's existence pretty-much proved) by the finding of a single useful locus in humans without parallel in any monkey or ape. (Failure to find such a gene, after a complete search, would not falsify progressive creationism, so these folks cannot lose.) I haven't heard of any "progressive creationist" undertaking such a search. Further, if "progressive creationism" is true, and God makes a practice of adjusting the genes in gametes, then what do we say to people with genetic diseases (birth defects, cancer) conferring no advantage? Why not choose a more stable system, and change it only for creatures' benefit? And Darwin's theory is called "cruel"? If you want to get in touch with the handful of active creationists who possess integrity, check with the American Scientific Affiliation or Students for Origins Research; nowadays these people mostly just remind others that the available data does not justify the naïve naturalism of, say, Carl Sagan. I look with hope to these people, especially as religious conservatives become aware of the facts. SOR and ASA folks will tell you more about the "progressive creationist" option, which I suspect most of the members hold. Maybe one day they'll report an experiment.... In Oct. 1997, the National Association of Biology Teachers had enough sense to revise its definition of evolution to remove the words "impersonal" and "unsupervised". This was past due, and something I'd worked for over the years.
* (5) Lately there's a minimalist approach to "human origins" from Religious Right types which doesn't even require the interventions (and the problems) of progressive creationism. "The Bible says only that the human race was specially created, not any other species". Because apes were so successful, God used the DNA sequence from one of them (junk sequences and all), very slightly modified, to make Adam by special creation. This idea is probably unfalsifiable and will appeal both to people who are committed to supernatural creationism and to people who are highly impressed with how different we are from all other animals.
* (6) You have yet another option that's consistent with the facts, and actually easy to believe. In 1983, I coined the phrase "The New Design Argument" for a common argument that begins with the idea that the Good Lord set up the physical constants for our universe so that the whole process leading to human beings (or whatever we're supposed to lead to) could happen without the Good Lord having to intervene at all (on our planet, and most likely on a billion others); this got some smiles even if it didn't end discussion (and I don't find it persuasive, either), but the idea was developed soon afterwards by an Anglican bishop (probably independently) in a soft-science book ("The Probability of God"). Much earlier, Pythagoras taught that the mystery of creation was placed by the Creator in the math. Spend some time with the new physics (quantum physics, relativity) and you may end up believing this.
* (7) The incredible complexity and beauty of the genetic code impresses many contemporary thinkers (especially non-geneticists) as proof of intelligent design. Before accepting the implications of this argument (the old "theological design argument", originally applied to individual species or genera, but now pushed back 3 billion years or so by "progressive creationists"), familiarize yourself with ribozymes (Science 231: 545, 1986; Cech & Altman Nobel prize 1990; Nature 352: 516, 1991; Nature 358: 543, 1992, Science 256: 1396 & 1416, 1992; Nature 374: 777, 1995) and natural selection (actually, Darwin's cycle of selection -> replication -> mutation -> selection) at the molecular level (Science 255: 800, 1992; Darwin's laws in the RNA world Science 257: 635, 1992; evolution in the test tube Nature 361: 119, 1993; this is now routine Science 267: 237, 1995; RNA world Sci. Am. 274(1): 27, 1996). We still have and use ribozymes (Nature 364: 358, 1993); it's amusing to think this is probably the original stuff. Synthetic self-replicating molecules: Sci. Am. 271(1): 48, July 1994. Likely origin of the genetic code, by analyzing huge numbers of tRNA sequences: Proc. Nat. Acad. Sci. 93: 452, 1996. Given present knowledge, I'd consider chemical evolution leading to the genetic code (though not the fact of our common ancestry with other animals) to be an open question; you may disagree either way.
* (8) The theory and practice of molecular phylogeny: Science 264: 671, 1994. The whole history of life in Darwin's world arouses in me the same sense of wonder and awe as does the sight of the heaven (Psalms 8 and 19 and 104 and so forth). My best answer begins with the observation that none of the great world scriptures focus on the Creator's role as Cosmic Designer (as certain more recent philosophers have tried to do). Instead, they address the more immediate concerns of human beings. Dennis Burkitt (of "Burkitt's lymphoma") was an evangelical Christian and he and I swapped letters on the subject. His analogy is the rider of a train; what matters is where you've going, not where you've been. We hear of various people who met the Good Lord, but never about their getting lectures on the mechanics of creation. John Paul II remarks that "the Bible doesn't tell how the heavens were made, only how to get there". Today's process theologians even talk about creation as a "planned accident" in which most creatures are pretty happy most of the time (see, for example, the best-sellers about "why bad things happen to good people", which are process theology by a thoughtful rabbi; good reading for any physician interested in religion, too.) Saying grace, I thank the Good Lord for our food, but I know where food comes from. We may thank the Good Lord for the birth of a child, but we know where babies come from; nobody demands "equal time for stork science." Charles Darwin actually pointed this out in the "secondary causes" passage in "The Origin of Species", in which he describes the Creator setting up the laws of nature and allowing species to have natural origins just as individuals do. In the Sermon on the Mount, I was told that the Good Lord feeds the birds and makes the lilies beautiful, but no one, then or now, could have understood this as literally as today's creationists would interpret Genesis. Later in the course notes, I'll defend my belief that a human being is a spiritual process joined, somehow and for some presently-unknown reason, and only temporarily, to a body that has arisen by natural processes.
* My best answer ends with the observation that (like Job, re-read the Old Testament book) I'd rather settle for less-than-complete explanations than believe the facile, obviously-wrong answers generated by uncharitable ignorance.
* (9) Left-wingers: Your "Gaia hypothesis", like "creation science" for your conservative counterparts, can be invoked to explain anything and has shown no predictive value whatever (Nature 345: 470, 1990.)

* It's a long way from Amphioxus / It's a long way to us;
It's a long way from Amphioxus / To the meanest human cuss;
Good-bye, fins and gill-slits, / Hello, lungs and hair,
It's a long, long way from Amphioxus / But we come from there!
-- Anonymous
(* Like it or not, it's the truth.)
Assortive mating: One of the distinguishing features of the human species. People do not mate at random, but choose partners who resemble themselves. This creates the remarkable diversity of individuals even within communities. As a result, there's somebody to do each of the complex tasks that maintain the community.
Non-paternity: In the words of the calypso song, "Your daddy ain't your daddy, but he don't know!" A sure way to confuse a geneticist (at least for a while.) Around 1% (Am. J. Hum. Genet. 51: 1171, 1992; the Sykes family tree Am. J. Hum. Genet. 66: 1417, 2000).
CYTOGENETIC DISORDERS
Aneuploidy results form failure of homologous chromosomes to move into separate progeny cells. This
may be from nondisjunction during the first meiotic division, or anaphase lag in any other cell division.
Rules:
(1)
Autosomal monosomy or no "X" chromosome causes early loss of the embryo.
(2)
All trisomies except trisomy 21 produce infants who will usually die during the first few months of life;
around half of early spontaneous abortions has a trisomy.
(3)
Unless a parent carries a balanced translocation, or when advanced parental age is a factor, there is no
real tendency for these problems to recur.
Beyond this, given the present limitations of our knowledge, the common chromosomal disorders
present a memory task for medical students.
Chromosomal breaks and rearrangements should be familiar to you.
Deletions indicate loss of part of a chromosome, either "terminal" or "interstitial".
Several curious birth defects are caused by microdeletions, i.e., loss of a few adjacent genes. As you
would expect, not all patients with these defects have the same phenotypes (why?)
Translocations are common; most are reciprocal translocations between two chromosomes. Unless
genes are damaged in the process, the patient is likely to be normal, but will produce lots of abnormal
gametes.
* "Robertsonian translocation" is a reciprocal translocation involving two acrocentric chromosomes,
producing a tiny chromosome that is lost and a very large chromosome.
* "Isochromosomes" result from faulty chromosome division. The products are a chromosome with two
long arms, and a chromosome with two short arms.
* "Inversions" involve two breaks in the same chromosome, with the portion between being re-incorporated backwards.
* "Ring chromosomes" result from deletions at both ends of a chromosome, with subsequent fusion of
their ends. Obviously, this chromosome is not going to take a normal role in mitosis.
There are several fragile chromosome syndromes (better, "chromosome instability syndromes"). "Big
Robbins" lists Bloom's syndrome (* small jaw, red blotches on face, short stature), Fanconi's anemia
(* gene cloned: Nature 356: 763, 1992), and ataxia-telangiectasia; there are several others. Not
surprisingly, these patients have high risk for cancer.
Trisomy 21: Down's syndrome (Lancet 361: 1281, 2003)
Although Down's syndrome is very common, we don't understand the reason that the extra chromosome
21 causes so many problems.
* We do know that the full expression requires the presence of 21q22. The genes for superoxide
dismutase, amyloid beta ("Alzheimer's amyloid" -- hmmm) and the oncogene ets-2 are all here.
It affects around 1 child in 700. Advanced maternal age is grave risk factor. Maybe 1 in 25 live births
to mothers over 45 have Down's. In only 20% of cases is the extra chromosome of paternal origin.
Most parents are cytogenetically normal, though occasionally one has a balanced translocation
95% have three separate 21's; 4% have a translocation; 1% are mosaics and less severely affected.
(Advanced maternal age is only a risk factor for the first group -- why?)
Pediatricians look for several signs. Don't expect to see them all:
{39121} Down's face
The condition is usually obvious at birth. Likely future health problems include:
Management of these children and young adults requires great understanding. Puberty compounds their
adjustment problems. Half are dead by age 30, but many make it to age 50 or beyond.
* Medical history buffs: Dr. Down, an Englishman of Victorian times, was among the first and most
ardent advocates of higher education for women. Neuroanatomy of Down's: Neurology 44: 1039,
1994.
Other cytogenetic autosomal problems
Trisomy 18 is Edward's syndrome.
Remember tiny jaw ("micrognathia"), overlapping fingers,
rocker-bottom feet, and cysts of the choroid plexus visible on
ultrasound before birth (beware: these last are by no means specific,
and are common enough in normal unborn children: Ob. Gyn. 90:
191, 1997).
{13450} trisomy 18; rocker bottom foot
Trisomy 13 is Patau's syndrome. Remember tiny head ("microcephaly"), arhinencephaly ("abnormal
limbic system"), tiny eyes ("microphthalmia"), polydactyly, and scrambled viscera.
Sometimes there is only one cerebral hemisphere ("holoprosencephaly") or even a single eye
("cyclops");
{11000} Trisomy 13
* Trisomy 9
{16604} Trisomy 9
Deletion of the short arm of chromosome 5 (i.e., 5p-) is
cat-cry ("cri du chat", "Is there a cat in
the
nursery?") syndrome. Children are usually
severely retarded
with severe behavioral problems,
but some are less affected and some survive into adulthood.
J Postgrad Med. 42: 86, 1996;
Arch. Dis. Child. 75: 448, 1996.
Velocardiofacial syndrome ("Shprintzen syndrome") was identified as a microdeletion of 22q11 in
1993. Because it is extremely common (about 1 person in 3000), it's worth remembering.
Kids generally have
More on velocardiofacial syndrome:
Am. J. Psych. 153: 1541, 1996; J. Ped. 123: 406, 1993).
* Deletion of 22q11 is the rule in DiGeorge's (Am. J. Hum. Genet. 51: 964, 1992). Isochromosome 12p
mosaicism (Am. J. Med. Genet. 47: 241, 1993) produces Pallister-Killian, etc., etc., etc.
Prader-Willi and Angelman syndromes: a major mystery of contemporary genetics. Read all about it
in Am. J. Med. Genet. 32: 285 & 514, 1989; 33: 66, 1989; 35: 319, 1990.
These two syndromes have been well-characterized for several decades. Each affects one child out of
a few thousand.
Prader-Willi patients are a little bit dull, typically have crossed eyes and almond-shaped epicanthic
folds, hypotonia (i.e, these are floppy babies), small hands and feet, growth delay, short stature, and
hypogonadism (low gonadotropins, not much puberty; some doctors give testosterone to selected Prader-Willi's, as in J. Ped.
114: 325, 1989). They overeat, incorrigibly stealing and hiding food, and become
very obese ("the commonest known cause of genetic obesity": NEJM 326: 807, 1992, no longer true
of course; "1 person in 10,000": NEJM 326: 1599, 1992, both good reading). Described as generally
docile and even "cute", they are also said to be prone to outbursts of extreme violence. (* Do you
remember the fat kid in "Full Metal Jacket"?) Major psychosis develops in many of these
people in early adult life (Lancet 359: 135, 2002.
* There's an albinism gene here, and the kids tend to be fair-complected (NEJM 330: 529, 1994).
Angelman patients, or "happy (?) puppets", are severely retarded, with microcephaly and huge jaws.
They have jerky, puppet-like movements, and laugh a lot (though apparently not in response to
pleasure). * The neuropathology has recently been described for the first time: Neurology 41: 416,
1991.
These two very distinct diseases are controlled by the same locus
(15q11-13).
Prader-Willi patients lack the normal gene from their father. A Prader-Willi child has inherited Dad's
mutant (typically deleted) gene, or else got two normal chromosome 15's from Mom and no
chromosome 15 from Dad (Nature 342: 281, 1989).
Angelman patients lack the normal gene from their mothers. So far, Angelman patients have inherited
Mom's gene deletion. (* Mouse gene at the same locus gives light pigmentation, and both types of kid
tend to be fair-complected: Science 257: 1121, 1992).
Puzzle that out! Parental imprinting of chromosomes is a hot topic, especially in the study of tumors.
* There's a familial Angelman, too, same locus; if the bad gene comes from Dad, the child is sick, if the
bad gene comes from Mom the child is normal; in other words, this version doesn't produce Prader-Willi. Am. J. Hum. Genet.
53: 140, 1993. The gene is now cloned, and has to do with ubiquitin-related
disposal of damaged proteins in the brain; it's only transcribed from the maternal chromosome: Nat.
Genet. 15: 70 & 74, 1997.
* News: Beckwith-Wiedemann syndrome victims (hypoglycemic, big tongue, asymmetric body, big kids
with a propensity for getting tumors) have both their 11p15's (IGF2 and WT2 are here) from their
fathers (i.e., the gestation was a trisomy, but a cell line discarded Mom's chromosome).
Am. J. Path. 154: 635, 1999.
* Common autism gene only affects you if you got it from Mom: Am. J. Hum. Genet. 60: 928, 1997.
* Note also that Wilms tumors, which lose alleles at the 11p13 position, generally lose Mom's rather than
Dad's. (Read all about it: Nature 351: 665, 1991; Lancet 338: 413, 1991).
* (Brachmann-Cornelia) De Lange syndrome: An important, relatively common (maybe 1:15,000 births;
there's a national organization) genetic disorder.
Affected children have small heads, hirsutism, a single bushy eyebrow, a small upturned nose, and a
down-turning upper lip. Many have deformed upper extremities. Most are very retarded, and most
succumb to infections early in life.
The genetics has eluded us so far. Current thinking suggests that cases represent new autosomal
dominant mutations, and that recurrences reflect mosaicism; a mother (no man has fathered two such
children on two different mothers) who has given birth to more than one such child often has "a mild
case of the disease"; there are now three dads who are mildly affected who've evidently
passed it on (Am. J. Med. Genet. 66:445 & 449, 1996. See Clin. Genet. 41: 42, 1992.
The recurrence rate if Mom has had a Cornelia de Lange baby
is about 3%.
{13404} Cornelia de Lange syndrome
Other microdeletion syndromes:
* Rubinstein-Tabyi (funny face, short thumbs, sometimes retarded) and Miller-Dieker are the prototypes.
In each, a few adjacent genes are lost. Wait for more.
Sex chromosomal disorders (* history of the sex chromosomes: Science 251: 1031, 1991)
Rules:
(1)
A Y-chromosome is necessary and sufficient to make a phenotypic male, provided the body can also
make and use testosterone. Well, usually -- it's actually the "testis determining
factor" gene that is usually present on the Y and usually not present on the X.
(2)
The more extraneous X-chromosomes, the more abnormal the person.
(3)
You will usually miss the diagnosis at birth, and may only make it late in adult life.
Klinefelter's syndrome
This occurs when a man has more than one X chromosome (i.e., 47,XXY, 48,XXXY, etc.). One man
in about 850 is affected. The etiology is unknown, but advanced maternal age contributes.
At puberty, the typical features generally appear. They include small testes, long arms and legs, often
smallish penis. Klinefelter patients generally are high-voiced, not very hairy, and (the big payoff!)
rarely go bald.
Because the Leydig cells do not function well, serum gonadotropins are high, Leydig cells are
hyperplastic, plasma testosterone is low, and (for some reason) estrogens are high, with about half
getting gynecomastia. The seminiferous tubules are always underdeveloped to some degree.
Many Klinefelter men have libidos and ejaculations, and many others don't. (The higher the testosterone
level, the more "maleness" and also, apparently, the higher the level of function and the more normal
the appearance: Abstract from Humangenetik 26: 61, 1975.) In any case, almost all of these men are
sterile, and Klinefelter's syndrome is a consideration whenever a couple is having difficulty having a
child.
XXY guys average lower IQ's than XY's, and psychologists talk about specific learning handicaps and
"diminished economic striving", but they are seldom retarded. XXY's are slightly over-represented in
prison populations, but the impact of the karyotype disappears when one controls for low IQ, and violent
XXY's are rare (Arch. Gen. Psych. 41: 93, 1984). Most Klinefelter's men are pleasant, easy-to-like
guys.
Around one male in 1000 has an extra Y chromosome. This is still "controversial" but won't be
resolved in today's political climate. On the average, these guys are taller
(Klin. Ped. 36: 39, 1997), have worse acne, have higher
average hFSH, hLH, and testosterone (this has held up nicely),
and allegedly average slightly lower IQ's
(this point's very questionable), than XY's.
When first discovered, it was hypothesized that XYY men would exhibit more
anti-social and impulsive
(i.e., "typically male" according to the ideology of the times)
behavior than other men (* popularized in the
"penal colony for XYYs" in Aliens 3). This
remains controversial.
One study (Arch. Gen. Psych. 41: 93, 1984) noted (1) these men
average substantially higher
testosterone levels, and are slightly over-represented in prison populations;
(2) among tall men of any
karyotype, the rate of conviction, especially for violent crimes, correlates surprisingly well with plasma
testosterone levels, with little additional contribution from karyotype; results of psychological tests
correlate poorly with all these variables; (3) the typical XYY's crime is
wife-beating; (4) the differences
between XYY's and their XY counterparts are anything but striking. (XYY's
found before birth are being followed, and this claim, which I made
in 1984, is now being confirmed: Prenatal Diagnosis 17: 363, 1997).
By the time you are ready for practice, perhaps we'll
know exactly what the extra Y does.
* The current work on XYY and mental/physical
problems is totally unimpressive --
small sample statistics ("five kids instead of three, out of 2000"),
anecdotes ("a single person with both XYY and schizophrenia";
"a single person with both XYY and teenaged lymphoma").
One group tried to quantitate the supposed angular facial features
of XYY men (Arch. Oral. Bio. 42: 579, 1997).
The one prospective study of Swedish children incidentally
discovered during a "let's screen all the new babies" fad
showed only a minor impact on behavior -- despite the fact that
their parents had almost certainly been told about the "criminal gene".
The study's "findings" result mostly from
the presence of a single
XYY
career criminal... and the authors couldn't find even one XYY who had
committed a crime with a weapon:
Psych. Med. 29: 953, 1999. Pretty slim pickings.
* Part of "political correctness" nowadays forbids geneticists
to "impose their own values" on parents. Before I set up my
XYY page,
I get 1-2 E-mails a
month from distraught parents who know they're going to have an XYY
boy "and the doctor wouldn't tell us the right thing to do".
I care more about people than about pseudo-ethics, so I'll
be straight with you -- I am satisfied that there is no
reason to abort a child for being XYY, or to be overly worried or
designate him as "special" or "different". Acne's treatable, and
tall is fun.
I think there won't be any serious work on XYY and
behavior in the near future.
In the meantime, Doc, PLEASE exercise caution before
predicting that "the extra Y will affect behavior".
Turner's syndrome
This is the result of monosomy for the short arm of the X chromosome.
About 1 out of every 2000 women are
affected.
Of these, around half are XO, and the remainder either have an isochromosome of the long arm of X,
or have partial deletion of the short arm of X, or are mosaics.
* Oddly, maybe 10% of spontaneous abortions would have been a Turner's, and 99% of XO conceptions
spontaneously abort (Nature 351: 406, 1991).
The major problem is failure of feminization at adolescence.
Patients have "webbed neck", "shield-shaped chest", and "cubitus valgus" (elbows turned out).
However, most patients are not diagnosed until the teens (if then). They fail to menstruate (i.e. "primary
amenorrhea" -- Turner's is the most common identifiable cause) or develop secondary sex
characteristics.
Almost all of the oocytes disappear by age 2, and patients have only "streak ovaries". As "Big Robbins"
puts it, "menopause occurs before menarche".
Rarely, lymph channels fail to form properly, and lymphedema of the hands and feet makes the
diagnosis apparent at birth. Or an alert clinician notes the "webbed neck" or "shield-shaped chest" of
the patient.
Another common problem in these patients is coarctation of the aorta.
* Psychologists talk about curious spatial perceptual problems in Turner's patients, etc.
* A male or non-XO female with Turner-like features has "Noonan syndrome". Some are XY/XO
mosaics; others have mutant PTPN11 (this is quite common, the commonest gene for congenital
heart disease. Update Nat. Med. 10: 849, 2004)..
{13433} Turner's, webbed neck
Multi-X females ("super-female")
Around one woman in 1000 has three or more X-chromosomes. Most 47,XXX women are normal, though
supposedly they are a bit slower than their sibs. The
48,XXXX woman are usually mildly retarded, and
49,XXXXX produces severe disability.
Hermaphrodites and intersex states: This is complicated. Learn these terms:
Genetic sex (really, chromosomal sex) is determined by the presence ("male")
or absence ("female") of the Y-chromosome.
The terminology needs changing now that we've discovered "testis determining factor", the presence or
absence of which should soon define genetic sex.
Occasional men have testis-determining factor gene, or something like it, on the X-chromosome, and
the dudes are 46, XX (J. Clin. End. Met. 76: 690, 1993, J. Urol. 149: 126, 1993).
Occasional women are XY's with a mutated testis determining factor gene (Hum. Genet. 88: 471, 1992;
gene is now called SRY, sex-determining region on Y: Nature 372: 525, 1994).
* Of course, these curiosities will run in the family. By the way, this is the answer to the old creationist's
objection "How could the mechanism of sex determination change?" The gene for maleness left the
autosome shortly after we branched off from duckbilled platypuses (Genomics 15: 317, 1993).
Gonadal sex is determined by the histology of the gonads, i.e., whether there are little eggs and follicles
(), or little tubules and Leydig cells (), or both (true hermaphrodite), or neither (?).
"Streak gonads" without the distinctive features of either sex are characteristic of Turner's syndrome
(XO people) and * Swyer's syndrome (XY people who have problems with testis determining factor or
its receptor).
Some guys have their testes vanish during fetal life ("vanishing testis syndrome"). They'll never be dads,
but hormone replacement turns them into fully-sexual men when the time comes.
Phenotypic sex ("genital sex" -- the use of the latter term may invite misunderstanding) is determined
by the external genitalia (male? female? can't be sure?)
True hermaphrodites, as noted above, have both ovarian and testicular tissue. Very, very rare. The
majority are 46,XX's with translocation of the Y to another chromosome, while most of the rest are
XX/XXY mosaics or XY's who have lost testis determining factor from a clone of cells during
embryogenesis (for the latter, see Am. J. Hum. Genet. 52: 578, 1993).
Pseudohermaphrodites have disparity between gonadal and phenotypic sex.
A female pseudohermaphrodite has a penis and ovaries, usually because of exposure to male hormones
before birth. The usual problem is a glitch in glucocorticoid synthesis, in which steroids are shunted into
male pathways. The "penis" is really a big clitoris (but aren't they all...?)
A male pseudohermaphrodite has a vulva and testes.
The infant may have a problem with testosterone biosynthesis, but normal sensitivity to testosterone,
so pubic and axillary hair develop when the "woman" reaches puberty. * Three such patients recently
proved to lack 5-α-reductase 2 (the enzyme that reduces testosterone to its active dihydrotestosterone
form in the genital area: Nature 354: 159, 1991).
Or the infant may lack testosterone receptors. This is testicular feminization (dumb name), a common
problem often inherited on the X-chromosome. These "women" usually have no axillary or pubic hair,
and of course, they don't menstruate. A sensitive article: Lancet 337: 33, 1991.
"Muellerian regression factor" ensures that neither type will have a uterus or oviducts, and the "groin
testes" should be removed because they tend to turn cancerous.
Don't confuse any of the above with gender dysphoria ("transsexualism" -- a boy insists he is a girl, a
man wants to be a woman or has even had a "sex-change" operation; less often, vice versa; Lancet 338:
603, 1991), transvestism (liking to wear the clothing of the opposite sex) or homosexuality/bisexuality.
Studying chromosomes and genes has contributed exactly nothing to our understanding of these states
of mind.
* The "conventional wisdom" has been to raise kids with ambiguous genitalia as girls (i.e., life is extremely
difficult for a man with a one-inch erect penis), and to operate early for cosmetic reasons. The wisdom
of surgery seems dubious, since at least some of these kids insist (without being told) that they are really
boys, and grow up into angry men. There are activists, etc., etc.
* In the 1970's, the Left
redefined gender to mean gender-role, i.e., your culturally-defined role as a male
or female, in order to emphasize the primacy of wicked cultural stereotyping over any biological
differences. If you read left-wing rhetoric, knowing this will help you understand it. ("Did you hear
about the postmodernist gangster? He made you an offer that could mean anything you wanted it to
mean!")
What's the real sex? Unless someone is a parent (the ultimate proof of maleness or femaleness), it all
depends on your definitions. Would anybody want to say that gorgeous --- ---, who supposedly has
testicular feminization, is "really a man"....?
INTRODUCING THE MENDELIAN DISORDERS
Enzyme defects cause substrates to accumulate (i.e., the storage diseases, alkaptonuria), and/or prevent
formation of a good end product (albinism, red hair, other white people) perhaps even with
accumulation of unwholesome precursors (Lesch-Nyhan), and/or fail to inactivate something bad (i.e.,
α1-protease inhibitor deficiency)
Defects in receptors and transport systems produce various malabsorption syndromes, urinary wasting
syndromes, unresponsiveness to hormones, problems mobilizing lipoproteins, and so forth.
Altered non-enzyme proteins include altered structure and function (hemoglobinopathies, collagen
problems), and abnormal quantities (the prime example is the thalassemia family)
Altered responses to drugs you'll study in "Pharm". If you lack G6PD, you get hemolytic anemia from
various oxidizing drugs, fava beans, and so forth. Some people are "slow acetylators" of certain drugs,
etc., etc.
AUTOSOMAL DOMINANT DISEASES
Rules:
When a person has only one good gene where most people have two, the person can expect to make 50%
as much of the good protein as do most other people. Sometimes, that isn't enough. Therefore, the
known autosomal dominant diseases fall into five categories.
(1)
Problems with the quantity or arrangement of large structural proteins
(2)
Problems with regulator proteins and receptors, that permit relatively good quality of life.
(3)
Deficiency in proteins that are in short supply even in health.
(4)
Anti-oncogene deletion syndromes, in which a "second hit" on the normal allele of a normal cell turns
it to a tumor cell. More about this last category later.
(5)
The mutant gene makes a harmful protein ("gain
of function"). Today, the best-understood of these are the prion-related
diseases, in which an altered protein begins a terrible chain reaction that can even be transmitted to
genetically normal creatures, even across species lines.
The common autosomal dominant diseases do not kill or disable until the patient has had a good chance
of having a family. Why? Hint: Most genetic diseases do not result from new mutations. The major
exceptions are Von Recklinghausen's neurofibromatosis and achondroplastic dwarfism (both are genes
with very high mutation rates).
The autosomal dominant disorders are mostly of variable penetrance and/or expressivity (why?).
Two doses of a bad autosomal dominant gene produces some severe exaggeration of the single-dose
syndrome, or else death in the womb.
Obviously, consanguinity does not play a role in autosomal dominant disease.
The major autosomal dominant disorders that you'll meet in this course:
Structural proteins (or the proteins that guide their arrangement):
Receptor / channel problems:
Short-supply protein deficiency syndromes
Tumor genes:
(including its variant Gardner's syndrome)
Harmful proteins / "gain of function"
The common thrombophilias (hypercoagulability problems, MUCH under-recognized):
Molecular biology unknown:
If you want, you could consider the common heterozygote semi-maladies "beta-thalassemia minor",
"sickle cell trait", "hemoglobin C trait", and "one-dose familial hemochromatosis" to be autosomal
dominant conditions. We won't argue.
The α-thalassemias are even more special, since the loci are double and the severity of the illness
depends on whether one has inherited one ("α-thal minima"), two ("α-thal minor"), three
("hemoglobin H disease"), or four ("hydrops fetalis") bad genes. Don't worry about these yet.
Marfan's syndrome
This is a heterogeneous group of genetic disorders with connective tissue problems. It is a physical
diagnostician's delight.
Marfan patients are tall, with very long extremities. The arm span exceeds the height. Joints are hyper-extensible (try
extending the thumb to touch the wrist; patients say they are "double-jointed"). The chest
is usually somewhat deformed, and the face may look funny. The bone structure is slim, the muscles
are wiry, and patients are generally slender.
The suspensory ligaments of the lens are often lax, and an ophthalmologist may diagnose "ectopia
lentis", which is almost pathognomonic. The globe is long and the cornea flat, so patients tend to be
very nearsighted.
The central portion of the thoracic aorta's media suffers breakdown of its fibers ("cystic medial
necrosis", a misnomer), creating a loose channel through which blood may extravasate ("aortic
dissection", kills about 1/3 of "true marfan people".
As the patient gets older, the aortic valve ring becomes lax, and the valve becomes incompetent. Severe
heart failure results. Marfan types also generally have the trivial "Barlow's mitral valve".
The most common Marfan locus is
a protein that is woven around
elastin (Nature 352: 279, 330, 334, & 337, 1991). * Congenital contractural arachnodactyly maps to
the fibrillin-like gene on chromosome 5: NEJM 326: 905, 1992). Variants and semi-marfan types abound -- around a hospital, string
beans like your lecturer (especially when he had TB and weighed 135 lb)
occasionally
get asked, "Are you a marfanoid?" (Probably not, unless you count all us skinny, nearsighted folks with
Barlow valves.) You'll need to decide for yourself whether the athletic, physically powerful, but funny-looking Abraham
Lincoln
could have been a "marfan" (seems less-than-classic), or had some variant
connective tissue anomaly (seems likely).
{13463} Marfan's, fingers
We can hope that finding the remaining gene(s) will further clarify the nature of marfanism.
In the meantime, there are two classical "models": (1) Lathyrism, caused by feeding sweet peas to
turkeys and resulting in fatal aortic dissection, results from beta-aminopropionitrile inhibiting lysine
oxidase, which cross-links collagen and elastin fibers. (2) Menke's kinky hair disease (* a rarity on the
X-chromosome), which prevents normal handling of copper, prevents function of lysine oxidase.
Of course, lysine oxidase structure and function, and the genes for collagen types I, II, and III are all
normal in Marfan's patients.
* Oddly, advanced paternal age seems to be linked to new Marfan's mutations. Stay tuned -- the study
of Marfan's syndrome may yield insights into the secrets of why we're built differently.
"Marfan variants" are numerous. (* I have friends from two different families with "probable Stickler's
syndrome"; one family is deaf, the other friend is a physician-athlete. Stickler's results from a premature
termination codon on the type II procollagen gene: Am. J. Hum. Genet. 52: 39, 1993; Am. J. Hum.
Genet. 53: 55, 1993.)
Ehlers-Danlos syndrome ("human pretzels") is a family of variably-inherited diseases that leave a
person with poorly-woven collagen.
Typical cases have overly-extensible (even "cigaret-paper") skin that gets hurt easily and heals poorly,
and very overly-mobile joints that often slip out of place.
* The following is presented only to give you an idea of the great variety of different problems that can
result in very similar clinical syndromes....
Type I and Type II: Classic Ehlers-Danlos, with lax joints and stretchy skin. Mutated type V collagen
(globby stuff that shapes and stabilizes type I collagen): Am. J. Hum. Genet. 60: 547, 1997.
Type IV: various problems with type III collagen ("reticulin"); colon and arteries often rupture
(review NEJM 342: 673, 2000).
Type VI: reduced lysyl hydroxylase (autosomal recessive), ruptured corneas, detached retinas
Type VII: inability to turn type I procollagen into collagen. Distinct but closely related "torn skin"
syndrome: Am. J. Hum. Genet. 51: 253, 1992.
Type IX: X-linked copper problem awaiting characterization, a copper problem
Familial hypertrophic cardiomyopathy ("sudden death during gym class", "Reggie Lewis's disease", etc.,
etc.) is due, in at least half of cases, to missense (i.e., single-base substitutions) in key
portions of the beta-cardiac myosin chain (NEJM 326: 1108, 1992). There are other
mutations (troponin, myosin binder: NEJM 338: 1249, 1998).
Familial hypercholesterolemia
"The most common mendelian disorder", with estimates of its frequency ranging from "Big Robbins"'s
conservative 1 in 500 to "minor forms" that maybe explain "why some healthy-living people run
higher LDL's than others". (Homozygotes with the really bad alleles die in their teens.)
To make a complicated story short (and extremely oversimplified), we think most of these patients lack
enough good apoprotein B-100 ("LDL") receptors. Therefore, they have trouble with:
(1)
hepatic clearance of VLDL leftovers ("IDL's") for recycling, leaving them in the plasma to turn into
LDL's
(2)
hepatic clearance of LDL's from the plasma, leaving high plasma LDL levels
(3)
receptor-mediated uptake of LDL's by other cells (do you remember "coated pits"?), leaving more
around to be taken up by the mononuclear phagocytes by their receptor-independent method.
You recall that receptor-mediated LDL uptake is tightly-regulated, but that non-receptor-mediated LDL
uptake is chaotic, and leads to lipid-bloated cells.
Dysregulated uptake of LDL's by macrophage clusters leads to "xanthomas", masses of lipid-laden cells.
Dysregulated uptake of LDL's by phagocytes of the arterial intima probably causes atherosclerosis.
The classic autosomal dominant disease features plasma cholesterol levels of around 300-500 mg/dL,
tendon xanthomas, and precocious atherosclerosis (with strokes, heart attacks, and so forth).
Classic familial hypercholesterolemia features 50% reduction in B-100 apoprotein receptor
effectiveness. This can be due to absence of receptors (most common), receptors that don't bind LDL,
or (rarely) receptors that don't work once LDL is bound. The genetics are more complex than cited above, and "familial high cholesterol" is clearly
heterogeneous. One gene ("for both high cholesterol and high triglycerides") has been found at 11q23-24 (apolipoprotein city;
Nature 349: 161, 1991). Stay tuned for the discovery of alleles.
The second attempt to cure a genetic disease using recombinant gene therapy was directed against LDL
receptor deficiency (in the Watanabe rabbit, of course). A retrovirus is used to introduce the good gene
into cultured liver cells, that are then re-introduced into the rabbit. This has now worked in a human
being (Science 260: 926, 1993; JAMA 269: 837, 1993).
* Other important, less-common lipoprotein defects are mutant apolipoprotein B-100 and mutant
apolipoprotein E; the latter produces the famous "type III hyperlipoprotienemia (remnant disease)". All
about genetic defects in lipoprotein metabolism: JAMA 265: 78, 1991. The apo-E deficient mouse:
Science 258: 468, 1992.
Stein-Leventhal syndrome is a mysterious very common woman's problem. The combination is:
Stein-Leventhal women also usually have:
Nobody understands Stein-Leventhal, but it appears to travel as a dominant
condition with variable
penetrance. You can treat it by manipulating hormones, and sometimes this seems to cure it. The male
phenotype seems to be the super-hairy guy who goes bald early. NEJM 333: 853, 1995; Clin. Encodr.
38: 653, 1993.
Since we haven't covered tumors yet, we'll look at anti-oncogene deletion syndromes under "Neoplasia".
Understanders: It is nearly certain that many "sporadic" birth defects represent "second hits" in people
inheriting a single autosomal gene for dysmorphism. A cell in the embryo is hit at the opposite, normal
allele, and a body part develops abnormally. One such case is known in the mouse (Ds; Am. J. Hum.
Genet. 52: 866, 1993). Finding these in humans (as the cause of birthmarks) is only a matter of time.
* Love may be blind, but it is evidently
not anosmic.
Since the HLA molecules combine with captured invaders to present
their antigens to the immune system, in an ever-changing world it's desirable
that people should have a variety. Of course, it's also good
to marry non-relatives. And indeed, people tend to marry people with different
HLA types more than you'd expect by chance. HLA type is an important
contributor to your individual musk, and in one study, women preferred
the dirty T-shirts of unknown men whose HLA types differed from them
(Proc. Roy. Soc. Lond. 260: 245, 1995).
This finding is now pretty robust (Am. J. Hum. Genet. 61: 494, 1997),
and the "electronic nose" (a computerized odor-detector) can tell your HL-A's
apart (Proc. Nat. Acad. Sci. 98: 9249, 2001).
AUTOSOMAL RECESSIVE DISEASES
Rules:
Many body proteins are in such abundant supply that if a person has only half as much of that protein
(i.e., has one good gene where most people have two), there is no obvious problem. However, if a
person has no good gene where most people have two, the person is sick. Therefore, the known
autosomal recessive diseases are either
(1)
deficiencies or defects in highly specialized proteins (enzymes, transport proteins), or
(2)
hemoglobinopathies requiring more than one dose of a gene
In contrast to autosomal dominant diseases, autosomal recessive diseases:
If the autosomal recessive disease is particularly common (worldwide, or in an ethnic group), it's likely
that the heterozygote state confers a significant advantage on its carrier. Examples:
Sickle cell and some other hemoglobinopathy carriers resist malaria.
Hemochromatosis carriers have much less trouble maintaining body iron stores.
Cystic fibrosis carriers are much
more resistant to gram-negative intestinal infections.
No one has a clue as to why Ashkenazi Gaucher's carriers have an advantage, but they must, since five
different mutations have gained a foothold (Proc. Nat. Acad. Sci. 90: 5384, 1993).
Here are the major autosomal recessive disorders that you'll meet in this course:
Deficiencies or defects in highly specialized proteins
Known proteins
Major hemoglobin problems
Carriers of most recessive traits can be detected by molecular biologists, who'll find, for example, "only
50% of normal enzyme activity", or "50% of the protein migrating abnormally", or "restriction fragment
length polymorphism".
Albinism: inability to synthesize melanin, our protective brown pigment.
Albinism involving eyes, hair and skin indicates homozygosity for defective genes at any one of twelve
or so autosomal loci. (* A form of albinism limited`(to the eyes is X-linked).
The best-understood form is deficiency in tyrosinase. The other ("tyrosinase positive") forms are poorly-understood, and
often form only part of complex genetic syndromes.
Regardless of the enzyme defect, patients with classic albinism suffer from light-sensitive eyes and skin,
skin cancers, and eye cancers.
{18253} albino
Alkaptonuria ("ochronosis"): lack of homogentisic acid oxidase
Homogentisic acid's nonenzymatic breakdown products ("alkapton", black stuff) accumulates in the
urine and cartilages (check those ears).
Articular cartilage wears out early, causing precocious arthritis of the spine and big joints.
Lysosomal storage diseases:
Lots and lots of these are known.
They result from failure of catabolism of large molecules within lysosomes. The molecular lesions
include lack of enzymes, mutant inactive enzymes, improperly-packaged enzymes, enzymes lacking
necessary activators or protectors, lack of helper proteins, or lack of proteins to move digested material
out of the lysosomes.
Tay-Sachs disease ("amaurotic familial idiocy"): lack of hexosaminidase A, causing accumulation of
GM2-ganglioside.
The prototype gangliosidase deficiency. Around one Ashkenazic person in 30 is a carrier. Neurons,
including those in the retina, are particularly affected.
"Ashkenazic" denotes an ethnic group, mostly of the Jewish faith, mostly from Eastern Europe.
Babies seem normal at birth, but become retarded, blind ("amaurotic"), uncoordinated, and limp. The
brain and head enlarge abnormally, due to accumulation of lipid. Death occurs within a few years. The
"cherry red spot" on the macula of the eye is actually the normal color, and the rest of the retina is too
white because of the lipid accumulation.
Light microscopy shows ballooned neurons, which ultimately die off. Electron microscopy shows
lamellar lipid masses in lysosomes.
{1321} Tay Sach's ballooned neurons
Natural selection at work: Tay-Sachs heterozygotes enjoy extra resistance to tuberculosis (Lancet 341:
214, 1993).
Niemann-Pick disease: lack of any one of several proteins required to break down sphingomyelin
molecules
Classic type A (no sphingomyelinase) features extensive accumulations of sphingomyelin and
cholesterol in neurons and the body's fixed phagocytes, notably liver and spleen. Patients die in early
childhood. By contrast, adults with type B have large livers and spleens but no CNS involvement
clinically.
Pathologists see lipid-laden, foamy-looking affected cells. Electron microscopy shows lamellar lipid
masses ("zebra bodies", other forms). * Medical history buffs: Pathology boasts two Dr. Picks: Louis
Pick described this disease, Arnold Pick discovered Pick's disease of the brain.
{20113} Niemann-Pick's in the liver
Gaucher's disease:
the genes Science 256: 794, 1992;
mouse Nature 357: 407, 1992): lack of glucocerebrosidase.
Type I Gaucher's disease ("adult type") results from subtotal deficiency of glucocerebrosidase. It is
very common in many communities, and one Ashkenazic person in twelve is a carrier. It is compatible
with a long, basically healthy life.
Patients have massively enlarged spleens (30 kg or more), and large livers and lymph nodes. Most of
these glucocerebrosides are probably from normal breakdown of old blood cells.
Patients develop pancytopenia (decreased numbers of circulating red cells, neutrophils, and platelets).
This reflects both bone marrow involvement and an overactive spleen ("hypersplenism").
Skeletal problems (bone pain, fractures) result from the marrow being packed with ever-expanding
cells. (For more about this see Orthop. Clin. N.A. 15: 765, 1984; Medicine 64: 310, 1985).
Pathologists see "Gaucher cells", huge reticuloendothelial cells bloated with glucocerebroside. Since
glucocerebroside is not lipid, affected cells are not "foamy"; instead, the good texture comparison is to
crumpled tissue paper. (* Old-timers compared it to "watered silk", which is hard to find nowadays.
Future pathologists: If you notice loaded cells in the alveoli, it's probably Gaucher's.)
Type II Gaucher's disease ("infantile type") resembles type I, except that the mutation is different
(NEJM 316: 570, 1987) and neurons are progressively destroyed (nobody knows exactly how, since
the storage is extra-neuronal). These children have progressive mental retardation and die after a few
years. This is not an ethnic disease.
Type III Gaucher's disease is of intermediate severity, and causes progressive dementia beginning in the
teenaged years.
* Roundsmanship: If you suspect Gaucher's disease but have no biopsy material yet, order a serum
L-tartrate-resistant acid phosphatase and a serum angiotensin-converting enzyme. Gaucher cells
elaborate lots of both.
* "Alglucerase", the replacement enzyme for Gaucher's disease, is available but costs $380,000 per year
(Proc. Nat. Acad. Sci. 90: 5384, 1993). Uh-huh. We learn with hope of the prospect of real gene
therapy: J. Hep. 30(S1): 1, 1999.
{00239} Gaucher cells, H&E
Problems degrading glycosaminoglycans ("mucopolysaccharides", such as heparan sulfate, dermatan
sulfate, keratan sulfate, chondroitin sulfate, and/or others). These include the very severe Hurler's
syndrome ("gargoyle" children with progressive mental retardation) to the variable Sanfilippo (severely
mental deterioration, near normal-looking) and Morquio (dwarves with bad aortic valves and normal
intelligence) syndromes.
Hunter's (MPS-II) is sex-lined, but all the others are autosomal recessives. Expect mild to severe
accumulation of mucopolysaccharides in the spleen, liver, etc.; * in some syndromes, the coronary
arterial intima is also progressively narrowed, leading to myocardial infarcts.
Pathologists see PAS-positive material in affected cells. Zebra bodies, which include lipid, may be seen
in normal lysosomes when the brain is involved.
{13413} Hurler's baby; abdomen protrudes because of large liver and spleen
* Medical history buffs: The brilliant Dr. Gertrude Hurler was one of the first female physicians in
Germany.
* This is as good a place as any to list the other inborn errors of metabolism that chiefly affect the brain.
Except as noted, they are autosomal recessives. Don't learn these just now; save them for later.
Metachromatic leukodystrophy: deficiency of arylsulfatase A; galactosyl sulfatide accumulates; brain
deteriorates after infancy. * Alleles: NEJM 324: 18, 1991.
Krabbe's globoid cell leukodystrophy: deficiency of galactocerebroside B galactosidase;
galactocerebroside accumulates; brain deteriorates in infancy.
Adrenoleukodystrophy ("Lorenzo's oil", etc.): a family of diseases, some X-linked, with problems
breaking down long-chain fatty acids; both white matter and adrenal cortical problems.
Glycogen storage diseases: The clinical application of a "Biochemistry" unit.
Type I (Von Gierke's disease, glucose-6-phosphatase deficiency): Patients have big livers and most of
the problems are due to hypoglycemia. A mild disease.
{07039} Von Gierke's disease, severe, with glycogen in the heart
Type II (Pompe's disease, lysosomal glucosidase deficiency, "acid maltase" deficiency): Patients have
involvement of all organs, and die young of heart disease. * A mild adult version exists (NEJM 315:
694, 1986).
* Type III (Cori's disease, limit dextrin disease, de-branching enzyme deficiency); Rare, patients have
liver storage problems. See Ann. Int. Med. 116: 896, 1992.
* Type IV (branching enzyme deficiency): accumulation of abnormal glycogen in all organs, including
the brain; death in infancy. Liver transplants for this disease: NEJM 324: 39, 1991.
Type V (McArdle's disease, muscle glycogen phosphorylase deficiency): Patients are poor athletes, and
get bad cramps and muscle damage when they try. Glycogen is deposited beneath the sarcolemma.
* Type VI (liver glycogen phosphorylase deficiency): Big liver, hypoglycemia, mild disease
* Type VII (muscle phosphofructokinase deficiency): Poor athletes, like type V; NEJM
324: 364, 1991.
* Type VIII (liver glycogen phosphorylase kinase deficiency): Big liver, hypoglycemia, mild disease
As you'd expect, these children store several different breakdown
products, hence "mucolipidosis". "I" refers to inclusions in the
cells of these children.
SEX-LINKED DISEASES
Chromosomes X and Y have only a short homologous regions at the tip of the short arm. (This is the
locus for pseudo-autosomal inheritance, seen only in uncommon disorders. This portion of X does not
lyonize.)
We await the definitive identification of any common genetic defect on the X-chromosome that is not
overridden by a normal allele on another X-chromosome (a real "X-linked dominant").
The best candidate for such a gene is one for manic-depression
(J. Med. Genet. 36: 585, 1999), but it remains unidentified.
The one well-known X-linked dominant disorder is familial vitamin D resistant rickets, an uncommon
phosphate-wasting problem. * Rett syndrome (MECP2) is autosomal
dominant. It causes a neurodegenerative
syndrome and a movement disorder (continuously wringing the hands)
beginning around age 5 months in affected girls
(Lancet 356: 830, 2000). Males with the allele die in utero or
have a much more severe encephalophaty.
* There are also a few exotic skin diseases that
are mostly lethal to the male fetus.
Most problems on the X-chromosome require one dose ("dominant" for hemizygous men), two doses
("recessive") for women.
Rules:
X-linked diseases:
The only two conditions known to be carried on the Y-chromosome are:
(1)
being a man ("testis determining factor": Nature 351: 325, 1991).
(2)
having lots of hair grow on your ears when you get old. Actually,
even this one isn't true (Eur. J. Hum. Genet. 12: 1077, 2004).
(3) Some mutations that render a guy almost completely infertile
(Gene 321: 25, 2003, Am. J. Med. Genet. 41: 814, 2004; others).
These are passed father-to-son.
We get our Y-chromosomes exclusively via the male line, just as we get our mitochondria exclusively
by the female line.
* Adam, our common male ancestor, seems to have lived in Africa (if you accept some straightforward
assumptions): Science 251: 378 & 379, 1991. Eve did too, around 135,000 years ago (Ph. Tr. Royal
Soc. London 337: 167, 1992). More about the recent African origins of contemporary humankind: Nat.
Gen. 13: 154, 1996.
* What's worse, it seems that the coalescence time for the Y's is considerably shorter than the coalescence
time for the X's. See Nature 378: 376 & 378, 1995. Any ideas how this could be? (HINT: "Like it
or not, we are the descendants of the men who won wars.")
* The Neanderthals apparently buried their children
(Nature 377: 585,
1995), practiced cannibalism (Science 286: 128, 1999), and
may have used the same musical scale as we do (Sci. Am. 277:
28, 1997). Right now it looks as if they were a separate species
(Nature 412: 534, 2001 morphology, Nature 404: 453, 2000), i.e.,
we
exterminated them (Am. J. Hum. Genet. 55: 760, 1994;
Am. J. Hum. Genet. 59: 185, 1996)
* If we're to believe one new study,
about 8% of men in the regions of Asia that were part of the Mongol
empire are male-line descendants of Genghis Khan (Am. J. Hum. Genet. 72:
717, 2003).
* Microdeletions on "Y" probably account for a few percent of cases of male infertility: NEJM 336: 534,
1997.
The major sex-linked disorders that you'll meet in this course:
Known proteins
Fragile X chromosome
Fabry's disease (* "angiokeratoma corporis diffusum universale"; deficiency of the enzyme
alpha-galactosidase A)
Ceramide trihexose accumulates in blood vessels and elsewhere; especially involved are the renal
glomeruli (look for big foam cells here). There is also some brain involvement,
and the peripheral nerve involvement makes this the most painful of the storage diseases.
* "Angiokeratomas" ("tumors of vessels with keratin") are rough, red bumps. Don't confuse these with
the smaller, similar-looking angiokeratomas that are common on the scrotums of many older men.
{07967} Fabry's, lamellar bodies on electron microscopy
Fragile X syndrome (* Martin-Bell syndrome):
see Am. Fam.
Phys. 39(5): 185, 1989; JAMA 271: 536, 1994 (deep stuff), or Am. J. Med. Genet. 30(1-2), whole
issue. Fragile X foundation: Box 300233, Denver, Colorado 80203. Screening: Br. Med. J. 305: 265,
1992.
Around half of cases of familial mental retardation now appear due to a single genetic defect, detectable
using cytogenetics, in which a defective gene at Xq27.3 presents as discontinuity seen on G-banding.
* Fragile X protein has to do with synapse morphology, and affected people have long-skinny dendritic
spines and small buttons (Proc. Nat. Acad. Sci. 94: 540, 1997).
The syndrome is almost as common as Down's; around 1 guy in 1500 has it. Affected men have IQ's
in the 35-50 range, and distinctively large testes (the only reliable physical finding, though there may
be other abnormalities: "big ears, big upper jaw, big testicles").
By the way, about a third of heterozygous women are also retarded (Am. J. Hum. Genet. 52: 884,
1993). It turns out the problem isn't lyonization, but the severity of the mutation (JAMA 271: 507,
1994). Deep stuff.
* Molecular biology: Nature 349: 624, 1991; JAMA 271: 536, 1994. Surprisingly, not every guy with
the abnormal sequence is affected; the defective DNA must also be tagged with methyl-cytosines
(Science 251: 1236, 1991). The gene that gets turned off is FMR1. Its product is FMRP, and you can
test for the absence of this thing using an antibody (cheapo; Lancet 345: 1147, 1995).
* Medical history buffs: Julia Bell, an English physician who characterized this illness, began her career
as a major astronomer.
In the future, look for other "fragile site" diseases. This may go a long way to explaining "polygenic
inheritance", "variable expressivity", spotted animals, etc. (Ideas: Lancet 338: 289, 1991).
{53764} fragile-X guy (long maxilla)
Lesch-Nyhan is already partly familiar to you from Biochemistry's discussion of gout, but this is only
part of the story of this grisly disease.
The neuropathology (i.e., why these kids chew off their body parts) has been clarified. There is a
profound lack of dopaminergic terminals and cells (NEJM 334: 1568, 1996).
MITOCHONDRIAL INHERITANCE
We are just beginning to discover diseases that are carried in the extranuclear DNA of the mitochondria.
Of course, we get our mitochondria, with their genes, only from our mothers. (* We believe
paternal transmission is vanishingly rare; first proven
case in a human NEJM 347: 576, 2002.) Obviously, a variable number of
mitochondria in various family members are affected, and the severity seems to depend (at least in the
case of myoclonus epilepsy; see below) on the overall % of involved mitochondria.
Important: All these diseases are progressive, and affect cells non-uniformly. Different cells bear widely
variable numbers of bad mitochondria. What is clearly happening is that the mutation gives the bad
mitochondria a growth advantage over their normal counterparts.
The first cloned mitochondrial disease gene was the one for Leber's hereditary optic atrophy (Science
242: 1427, 1988; Sci. Am. 259(4): 32, Oct. 88; NEJM 320: 1300 & 1331, 1989; Am. J. Hum. Genet.
51: 378, 1992; another problem with cytochrome oxidase); progressive visual impairment. The name's
worth remembering because it made history.
Soon afterwards, it became clear that most cases of Kearns-Sayre disease (progressive external
ophthalmoplegia, retinal pigmentation, heart block, cerebellar ataxia) and progressive external
ophthalmoplegia (without the other stigmata of K-S diseases) were found to be caused by various
deletions of the mitochondrial genome (NEJM 320: 1293, 1989; Science 244: 346, 1989).
Mitochondrially-inherited illnesses are progressive, i.e., not apparent at birth, but eventually causing
disability. Every mitochondrion, of course, has many copies of its genome, and probably the "bad"
loops somehow overgrow the "good" loops over the course of time. For more on this idea, see Am. J.
Hum. Genet. 48: 649, 1991.
Myoclonus epilepsy with ragged red fibers is inherited maternally. The cause was found in 1990 to be
due to a mutation in the lysine transfer RNA gene (Cell 61: 931, 1990; see also Lancet 2: 1253, 1988);
the "ragged red fibers" look that way because of proliferated, dysfunctional mitochondria packed around
their edges.
* MELAS ("mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes") results
from a disastrous mutation in a leucine transfer RNA gene (Nature 351: 194 & 236, 1991; the mutation
Proc. Nat. Acad. Sci. 89: 4221, 1992; a variant: Lancet 338: 143, 1991.)
* More: a deafness-and-diabetes mutation on tRNA(Leu(UUR)): Diabetes 43: 746, 1994.
Other "mitochondrial diseases" (notably the other "mitochondrial myopathies with ragged red fibers"
and Leigh's) are coded (at least in most cases) by the nuclear DNA. * Pearson's: J. Clin. Invest. 86:
1601, 1991. * We are just now linking particular tumors to mutated mitochondria DNA (more about
this under "Kidney").
All about the mitochondrial genome in health: Ann. Rev. Cell. Bio. 4: 289, 1988. Maternal inheritance
is a near-synonym for "mitochondrial inheritance", but you must rule out an infectious organism that
can only be passed from person to person across the placenta. ("* But mitochondria are really...."; see
Nature 364: 358, 1993. Nobody questions nowadays that cyanobacteria gave rise to chloroplasts, and
the biologists tell us that mitochondria sure seem a lot like purple bacteria.)
* Wolfram's disease, an autosomal recessive disease on chromosome 4, causes loss of DNA from the
mitochondria. Nobody knows how. J. Clin. Invest. 97: 1570, 1996.
POLYGENIC DISORDERS (much better term than "Big Robbins's" "multifactorial inheritance")
Diseases and conditions in which heredity clearly plays a dominant role, but for which there is clearly
no single gene, include:
Telling polygenic disorders from "single-gene diseases with incomplete penetrance" or "genetic diseases
modified by environment" or "genetic heterogeneity" is a tricky business. Evidence for "polygenic
inheritance" of a particular disease includes:
(1)
Adoption studies demonstrate that "nature" is more important than "nurture" for this disease.
(2)
There is no classic pattern of inheritance in most large kindreds.
(3)
Parents and sibs are at some (approximately the same) increased risk;
(4)
The sicker the index patient, the greater the risk to the near-kin;
(5)
The more sibs that are affected, the greater the risk to the next baby;
(6)
Diseases are of variable severity, and minor or even trivial cases can be found.
(7)
Because the disease is caused by "piling up of several abnormalities", environment modifies the full
expression of most of these diseases, and identical twins of patients, while at high risk, are not invariably
affected. (The closest we have to an exception: Type II diabetes)
Heredity obviously plays a role in height, build, looks, coloration, voice, and masculine hair
distribution-loss pattern. Most people would admit that intelligence ("defined to be what is measured
by IQ tests", sorry; despite ideology see Nature 367: 591, 1994), personality (whatever that is), and
tendency to gain fat (i.e., appetite), along with certain aptitudes, depend at least in part on one's genetic
background.
Our knowledge of the actual genes is minimal. Be extremely skeptical about "scientific studies" of such
things, especially when race is an issue, or the writer sounds very confident.
More plausible are studies that find a single locus that seems to modify several "distinct polygenic
diseases" (a "Beavis and Butt-Head" D2 dopamine receptor variant that correlates with nastiness: JAMA
266: 1793, 1991; Arch. Gen. Psych. 49: 157, 1992).
Right now, watch the genetics of obesity -- it's probable that many, if not most, overweight folks have
a defective leptin receptor in the brain (NEJM 332: 679, 1996), its expression modified (probably) by
other genes.
GENETIC DISEASES THAT ARE NEVER INHERITED
Sounds impossible, but isn't. Rules:
1. A fertilized egg with the disease is non-viable.
2. All patients with these diseases have
a post-zygotic mutation involving only
some of their cells, and express their conditions variably depending on where
affected cells may be.
There's at least three so far:
Stay tuned for more.
By the way: Creutzfeldt-Jakob's (prions) is a disease that produces an infectious particle that can be
propagated to healthy people. There is a genetic subtype that generates infectious, transmissible
prions.
By the way: Neoplasms are the great acquired genetic disease. More about this soon.
* GENETIC DISEASE IN CONTEXT
In response to recent press coverage of advances in genetics, you [the journal Nature] are quick to point
out the imagined political and social dangers of a belief in biological determinism, citing Nazi
Germany
as a precedent (Nature 387: 743, 1997). Perhaps you could explain to your readers why similar
comments, quoting the vastly greater number of people who perished at the hands of regimes committed
to the dogma of cultural determinism in the Soviet Union, China, Cambodia and elsewhere are never
made.... The power to kill millions during our century has come much more from the belief that human
beings are the hapless victims of "ideology", "society" or "class" than it has from any knowledge of
genetics, however faulty or misapplied it may have been.
-- Christopher Badcock, Nature 388: 13, 1997
Every physician is probably interested in at least one genetic disease. We must offer sound guidance
both to individuals and to society, and the philosophical and emotional issues raised by genetic disease
fall within the proper range of an introductory pathology course.
First of all, in counseling individuals, especially about decisions that involve terminating a pregnancy
or foregoing parenthood, you must obey the law and be considerate of the feelings and priorities of
others, which may be different from your own. (Sensitive review: Lancet 338: 998, 1991.)
It's obvious to you that carrying a genetic problem is not the person's "fault". But remember that even
today, many people are unaware of this. Address these concerns.
And please don't call children with genetic syndromes (known or unknown) "FLK's" ("funny looking
kids") or "GORK's" ("God only really knows" what's the matter). Try to say "dysmorphic", instead.
During the 20th century, the two horrible
totalitarian systems differed fundamentally
on their concepts of nature-vs.-nurture. The Nazis believed (or pretended
to believe) that you were
good or bad depending on your race, i.e., for the Nazis, genetics
was everything. The Communists
believed (or pretended to believe) that they would change human nature itself,
and make everybody good, by passing laws that forced people to share. For
the Communists, environment was everything. Both are obviously wrong, both
in the theory and in its application. It is equally wrong today to deny
either the impact of genes, or the impact of environment, on health
and behavior.
Genetic testing without genetic counseling is malpractice. But no matter how well you counsel people,
bear in mind that a large minority of them will grossly misunderstand what you have told them (review:
JAMA 263: 2777, 1990). Is this a reason to forego attempts at educating the public? The author
suggests "yes"; I am not sure I agree.
Most people, including most people who lead public opinion do not understand today's biology. Your
neighbors are frightened by today's talk of gene splicing, gene therapy (update
Proc. Nat. Acad. Sci. 94: 12744, 1997, also
NEJM 333: 871, 1995; gene therapy of cystic fibrosis in humans begins Nature 362:
450, 452, & 453, 1993; and a mouse is now cured of cystic fibrosis Nature 362: 250, 1993;
gene therapy without red tape in Mainland China: Nature 367: 1,
gene therapy review: focus on ovarian carcinoma
Am. J. Clin. Path. 109: 444, 1998.
1994), etc. The public understands when
cancer chemotherapy
kills people by the thousands each year. Gene therapy was
set back by the death of ornithine transcarbamylase deficiency victim Jesse
Gelsinger (late 1999) in a gene therapy experiment conducted by
a physician with a strong financial interest in the outcome, and who had
some ethical lapses.
More fears are fueled by B-movies (did you ever see a movie or a TV series in which the
hero was a true-to-life, non-weird scientist or engineer?), by activists (both "liberal-green" and
"conservative"), by press coverage (still generally negative), and by some obvious (even when
"politically correct") charlatans (Nature 303: 563, 1983; Discover Jan 1985, p. 34; Science 233: 704,
1986; Discover June 1986, p. 50 ff; Forbes 141: 138, June 27, 1988; Nature 354: 779, 1991;
Sci. Am. 272(2): 29, Aug. 1997.
The
world's leading anti-biotechnology activist (Jeremy Rifkin, a left-wing lawyer who
has made a career manipulating the legal system
in order to delay or block bioengineered products such as
medicines, vaccines, and high-yield food crops) appeared in 1989 on
Public Radio, objecting to digital watches. He complained that they do not have hands that simulate
the daily rotation of the earth, and will therefore cause us to care less about the problems of the
environment.
One current nutty-Left idea is that all "scientific knowledge" is merely a construct designed
to further the political agendas of the scientists, see Science 255: 613, 1992, Paul Gross's 1993 book
"The Higher Superstition: The Academic Left and its Quarrels with Science", etc., etc.; traditionally,
left-wing screwballs since Karl Marx have abused terms from academic philosophy and today they're
calling all their anti-rational, anti-scientific stuff "postmodernism" (Science 261: 143, 1993,
http://www.pathguy.com/postmod.htm). As gene therapy proves successful in making sick children
healthy, I would like to rub all these peoples' faces in it. I am not making any of this up. However, the
issues are serious.
Recombinant DNA techniques make possible the production of contaminant-free forms of growth
hormone, anti-hemophilic factor, interferons, and tissue plasminogen activator, as well as the best
insulin and dozens of other medicines. We can even improve food safety and availability (JAMA 269:
910, 1993). But the public still talks about "human beings tinkering with life" and "doctors creating
new epidemic diseases in the lab". The real bunko artists also talk about "species integrity" (Science
News Nov. 3, 1984, p. 278) and "pure food" (JAMA 269: 910, 1993, Nature 361: 6, 1993, others), and
some contemporary "green" folklore still attributes the origin of AIDS to sloppy recombinant DNA
work. Your patients are reading this junk.
Recombinant DNA techniques offer vast improvements in early detection of cancer, prosecuting murder
and rape cases, proving paternity, identifying remains (J. For. Sci. 38: 686, 1993: you can get good
DNA from brain for years after death; J. For. Sci. 38: 542, 1993: identification of bones from the
Vietnam war; Forens. Sci. Int. 56: 65, 1992: how we identified the bones of Herr Doktor Josef
Mengele), and exonerating men falsely convicted and imprisoned for rape (Science 256: 301, 1992, not
to mention KC Star March 13, 1993. There are many such men. Guys: do you know how difficult and
expensive it is to defend against a bogus accusation of a sex crime?) But for many people, this simply
amounts to "new methods for invading our privacy" (Science 249: 1368, 1990).
Techniques for detecting carrier states have rendered several dread diseases completely preventable.
Yet this kind of work is widely perceived as "finding ways to discriminate against the handicapped".
(Not one of the horrid predictions of "advocates for the differently abled" have come true in the case of
α-1 protease inhibitor deficiency, which has been known for decades: JAMA 271: 217, 1994). But
there's still some reason to worry; "eugenics" has a mixed, often unsavory history, mostly because
pseudoscientists have written about it, and the "need for eugenics" (which, believe me, the public
understands) has been used to sell totalitarian and even racist politics.
Interestingly, there is now
talk about physicians having a duty to tell the
relatives of somebody with genetic disease that they
might have it, too (Ac. Med. 73 962, 1998 -- the author
says in no uncertain terms that this is too important to leave
to the "ethicists and lawyers").
Curiously, it is legal under the
Americans with Disabilities Act to fire somebody for carrying, but not being sick with, a hereditary
disease (Sci. Am. 270(6): 88, 1994). The same foolishness
in reverse: A major medical journal (Lancet
343: 583, 1994) complains that "Tibetans are victims of racial dilution", meaning that those awful Red
Chinese allow a young person in Tibet to marry whoever he or she loves.
Historians see Am. J. Hum.
Genet. 49: 1109, 1991.
In contemporary Mainland China,
screening is practiced as in the U.S., but parents are forbidden by law from bringing a fetus known to
carry a dread birth defect to term. The free world's outcry against this policy: Nature 367: 1, 1994.
This sounds to me like selective indignation on a planet on which 50,000 healthy people, mostly
children, die daily of malnutrition. You may disagree.
Whatever you may think about this, it is likely that several countries will
follow Mainland China's politically-incorrect lead by trying to control genetic disease by regulating fertility,
creating a host of new problems. And at least a few scientists are now asking, "Can we afford not to
engage in eugenics?": Nature 353: 598, 1991. This provokes howls of protest. "Slippery slope"
thinkers: Am. J. Hum. Genet. 51: 222, 1992. The famous "slippery slope" argument (fallacy?)
reviewed: J. Med. Eth. 19: 169, 1993 (finally somebody has the guts to say that it's demagoguery
simply to shout "That could lead to Nazi-style genocide!" every time somebody talks about limited-resources or
death-with-dignity or whatever).
A disgusted academic geneticist finally puts the
problem in perspective, and asks why contemporary "gene ethicists"
constantly carry on over "justice
and the human genome project" or "let's talk about Hitler's atrocities again" and never mention the
population problem or its true cause, the real and genuine
oppression of the poor (Am. J. Hum. Genet. 56: 538, 1995).
It is hard not to be moved by the plights of teenagers with sickle-cell disease, hemophilia, or cystic
fibrosis. Of more concern to politicians, the expense is tremendous. A hemophiliac consumes an
average of about $60,000 in health care each year. An institutionalized
retarded person consumes $130,000 / year.
Screening programs for Tay-Sachs disease (Ashkenazi people) and beta-thalassemia (Mediterranean
people) have substantially reduced new cases of these diseases (Thorax 45: 46, 1990). The situation
with sickle cell disease is complex. However, scientific illiteracy in the U.S. is massive. It is now
commonplace for "carriers of genetic disease" to lose their health insurance and/or jobs (NEJM 323:
62, 1990; Am. J. Hum. Genet. 50: 457 & 465, 1992); only in 1996 did Bill Clinton sign into a law a
prohibition on treating genetic predisposition as a "pre-existing condition" for which coverage is denied.
Ethics of genetic research (sane article: Nat. Genet. 15: 16, 1997).
Linus Pauling, who worked out the biology of sickle cell disease, wrote very harshly about people who
knowingly bring a sickle cell child into the world (UCLA Law Review 15: 268, 1968), and it is
probably only a matter of time before a child with sickle cell disease sues his parents for negligence (J.
Leg. Med. 5: 63, 1984). Left-wing apologists for the Brooklyn Museum
of Art's 1999 exhibition of "art" created from genuine animal feces and
depicting the Virgin Mary surrounded
by weird genitalia defended it on the grounds that it warned
of the dangers of genetic engineering.
In the late 1990's, pressure groups
climbing to speak for the world's "indiginous peoples"
called for a halt to the Human Genome Project. Among other reasons,
it was giving results that contradicted some of
their mythologies (I use the word
without apology), and they were concerned that people with a remote
ancestor might be entitled to share their affirmative-action benefits
(Am. J. Hum. Genet. 63: 673, 1998; Am. J. Hum. Genet. 64:
1719, 1999). The word that these people have coined is "biopiracy".
So far as I can tell, the search for truth goes on anyway.
If you're interested in philosophy in ethics, you'll notice
that the current orthodoxy in discussing "ethical
problems" in eugenics considers autonomy to be the supreme good, and that "human rights"
includes the right to bring as many profoundly crippled and
profoundly unhappy children
into the world as you want. This is probably in reaction against
the sometimes-unsavory history of "eugenics" many decades ago ("Remain non-directive; simply tell
them the risks" was framed in the Nuremberg / Little Rock era.) Or, heaven forbid... the ethicists might
actually be afraid of the mudslingers. As a doctor, I can (and probably should)
tell a person, "You have no business leaving that melanoma on your face", or "So you're a Jehovah's
Witness? I'll take you to court to get a blood transfusion into your child who is dying of hemolytic
disease of the newborn." However, I must never, ever say (unless explicitly asked, and then I must be
oh-so-careful), "In my opinion, you two have no business having another kid with Lesch-Nyhan
syndrome." Again, to be frank, this perplexes me, and I suspect it perplexes other folks, too. For the
current ideology, see, for example, Am. J. Hum. Genet. 54: 148 & 159, 1994. (Tying the tubes of
chronic mental patients who were totally incapable for caring for their babies was horribly wrong
because most of them were poor and this is "classism". The desire and ability to have healthy children
will make us care less about sick children. "We now know that poverty can be solved through economic
and educational programs." That's a verbatim quotation, I didn't make it up. And so forth.) Am. J.
Hum. Genet. 60: 40, 1997 studied exactly how "non-directive" (i.e., virtuous) various genetic
counsellors are, and (read between the lines) seems to be (ssshh!) thinking the same things I am.
Today, antisense DNA technology ("magic bullets"; Science 260: 1510, 1993; Science 261: 1004,
1993; Nature 372: 333, 1994; delivery Proc. Nat. Acad. Sci. 93: 316, 1996) offers the reasonable
prospect of really effective biologic treatment of cancer (a mouse gets a leukemia remission from anti-BCR/ABL: Proc. Nat.
Acad.
Sci. 91: 4559, 1994), gene therapy offers the hope of effective treatment
of such dread diseases as muscular dystrophy, militants demand "more research to find a cure" for
lifestyle-related disease, while other people aggressively demand "up-to-date" lab techniques that do not
require the use of animals. Yet European "environmental activists", calling for a ban on all
biotechnology ("Nazi science") have recently gained great political clout, and their own militant wing
fire-bombs the labs of those who speak out in support of biotechnology (Science 255: 524, 1992). Most
"environmental activists" are more reasonable, but the Unabomber is typical of an important nut
movement who doesn't get much unfavorable press coverage. At present, there is almost no human
genetics or gene therapy research performed in Germany, since it is so easy for the
Left to make
political capital by comparing this kind of work to "Nazi medical atrocities". I am not making this up
(Science 264: 653, 1994). In 1995, a "green" leader in the German legislature said "We oppose any
research in human genetics", because "it undermines the dignity of human beings". See Nature 378:
437, 1995. In 1997, James Watson told Berlinners to "put Hitler behind us"
(Science 276: 892, 1997).
By contrast, the U.S. left-wingers
who framed the original 1996 Oakland "Ebonics" resolution
claimed it was proved that U.S. black speech is "genetically determined" and therefore not a dialect of
English but a fundamentally African language; they had been reading some pseudoscience. I am not
making this up, either.
When scientists suggest that it may someday be possible to replace the cystic fibrosis gene in a fertilized
egg (or a patient's somatic and germ cells) with a version that works, two objections are now routine:
(1) Pseudoscientists complain that "genetic diversity is necessary to ensure the survival of the species."
You'll recognize the misapplication of an important principle -- but could you make a Congressional
panel understand why it's wrong?
(2) Other people complain that repairing genes would "change who you are, violating your innate
personhood and unique individuality". Note that it is not quite right to say that "your genes make you
a unique individual" -- if this were true, then identical twins would be the same person, human cells in
tissue culture would enjoy full civil rights, and mosaics and chimeras would be two different people.
And complaints about "doctors going against the divine plan", dormant in most circles since the
anesthesia controversies of the late 1800's, are now being heard again. Incredibly, in 1983, leaders of
most of the large U.S. denominations (again, both "liberal" and "conservative") signed an inflammatory
document from Mr. Rifkin condemning all attempts to eliminate defective genes from the human germ
line (Nature 303: 563, 1983; we could have this underway in our lifetimes: Science 253: 841, 1991;
Science 262: 533, 1993; the first ones so treated will be anti-oncogene deletion syndromes).
Afterwards, several of them admitted they did not really agree with the document, but "wanted to
promote discussion" (huh?! did you forget about the 9th commandment, Pastor?), and more recent talk
from church circles has been much more honest and sensible (Hastings Center Report 14(2): 13-17,
Apr., 1984; Thomist 51(3): 501-20, 1987; Christianity Today 30(2): 22-28, Feb. 7, 1988; the Pope on
human reproduction, a moralist who's working on his scientific underpinnings: Nature 373: 100, 1995).
Thankfully, there now appears to be a consensus among all "reasonable parties to the debate" that
introducing genes into non-germline cells is ethically acceptable (J. Med. Philos. 16: 587, 1991; Nature
361: 5, 1993, gee whiz folks). Certain prominent members of the conservative religious-right (various
world-faiths, summer, 1995) produced another inflammatory
Rifkin document against genetically-engineered
creatures (Br. Med. J. 310: 1351, 1995); apparently it's fine to eat a cow but "abhorrent" to produce a
knockout mouse for cancer research. The next few decades will debate "germline enhancement",
the final decisions will be made by consumer demand, and the world will be healthier.
In particular, behavioral genetics asks questions (and is beginning to provide answers) that are certain
to upset people (Science 257: 164, 1992; conference canceled for reasons of "political correctness":
Nature 358: 357, 1992). "Which D4 (dopamine receptor) do you have?": Nature 358: 109 & 149,
1992. It is now generally accepted that D4 has a big effect one one's personality,
especially novelty seeking (J. Hum. Genet. 46: 26, 2001).
A claim about a D2 dopamine receptor as predisposing factor to alcohol and cocaine addiction
(Science 263: 176, 1994) flopped (Science 264: 1693, 1994).
By now, a host of animal models for personality -- especially aggressiveness --
are known, and present plausible biochemical mechanisms, and the same must
exist for humans based on what we know from adoption studies (Psych. Clin.
N.A. 20: 301, 1997.)
Some of these concerns are obviously
frivolous, just the Right or Left trying to make
political capital.
Others are not. Even complaints about curing genetic diseases raise questions about
possible uses in the remote future (Nature 312: 408, 1988).
I would cite history to argue that, when given the real facts, democratic societies almost always make
the right decisions. But many people will disagree (though their counter-arguments against science are
more likely to be based on some concept of human nature or disturbing anecdote rather than a major
historical trend).
Because of fears of creating monsters, most U.S. work with recombinant DNA was banned in 1975 and
remained very difficult for several years. In retrospect, this seems silly. (Frankly, ludicrous. In the
1970's, Mr. Rifkin's left-wingers disrupted biotechnology meetings by painting swastikas, etc., etc. Left-wingers:
doesn't painting swastikas count as "hate speech"?) For a contemporary account of "environmentalists"
who imagine that treating strawberries with a pseudomonad possessing a single inactivated gene would
prevent clouds from forming over California, see Nature 350: 284, 1991). One scholarly book, The
Gene Splicing Wars (AAAS, 1986), begins with the story of Chicken Little. Mr. Rifkin is "Foxy Loxy".
For the history of the Left's successes in delaying
bioengineered medicine, see
Science 230: 1146, 1985; Science 233: 516, 1986; Science 235: 159,
1987; Science News June 7, 1986, p. 366; Science 239: 341, 1988; Science 243: 734, 1989; New
Scientist 122: 29, May 27, 1989; Science 246: 30, 1989; Nature 337: 398, 1989; Forbes 144: 10, Oct.
2, 1989; Nature 346: 787, 1990; Nature 354: 257, 1991; Nature 358: 529, 1992; Science 251: 608,
1992. In Europe, it is now fashionable for left-wingers to
destroy fields of bioengineered crops ("Frankenstein food") at night.
The example I've heard most often is this: "The superoxide dismutase
genes is harmless in fruit flies, but it causes neurologic disease in humans.
This shows the danger of transferring genes from one species to another.
So there is no predicting what will happen."
Obviously, this relies on confusing alleles with loci, and anybody
who can't see the fallacy shouldn't be talking to the public
about science -- but they are.
Could you make a "Green" politician want to understand why it is wrong?
The flap over American beef and dairy products, which is probably
fueled by economic protectionism, is done under cover of green-party
"concern over safety"; in today's hungry world, real
humanitarians might instead
appreciate that hormone treatment can get cows to give 20% more milk.
Part of the irony is that genes recombine all the time in nature
-- in Mr. Rifkin's gut as well as at the NIH.
We learn with hope that political barriers to gene therapy have now pretty much disappeared in the
wake of its triumphs (Science 258: 744, 1992), at least in the U.S. (which is doing almost all of it).
However, the future promises to be thorny.
More troubling (for me, anyway) are the moral, ethical, and religious concerns raised by terminating
human lives (before birth, passive euthanasia of defective infants).
Most adults with whom I've discussed the matter express a preference for death over profound mental
or physical disability, and the law generally recognizes their right to refuse treatment. A few patients
want their lives preserved at all costs. Babies obviously cannot exercise this choice, and many
thoughtful people think we should assume that each would choose prolongation of life at great cost, or
that motives must be "purely unselfish" in dealing with these questions. Again, I am not sure I agree.
I would question the integrity of thinkers "on either side" who cannot see how aborting a fetus with
Duchenne's muscular dystrophy is like, and unlike, the atrocities of the second world war era.
The Human Genome Project, the task of sequencing the entire
human genome, is now complete,
thanks largely to
private corporations getting into the act (JAMA 279: 1933,
1998). It has already made the search for disease genes
much quicker (Br. Med. J. 314: 126, 1997).
Reviews: JAMA 280:1532, 1998, Arch. Neuro. 55:
1287, 1998, Am. J. Pub. Health 86: 1701, 1996. Francis Collins: Academic Medicine 73:
1241, 1998. Ethicists "expressing their concerns":
Lancet 352: 1448, 1998.
During the next few years, you will be hearing a lot more about "antisense" nucleic acids, synthesized
and introduced into cells to bind complementary mRNA and thereby inactivate the expression of
unwanted genes (i.e., oncogenes). The first major triumph has been keeping tomatoes ripe longer
(lawyers for the crackpot wing of "environmentalism" kept these off the supermarket shelves for several
years before their release in 1994, after which they held "tomato squashes" for
the media).
Today's genetically engineered crops include soybeans that require much
less bug-killer -- and today's "environmentalists" (who you'd think
would appreciate the need for less bug-killer) are now going gah-gah over the
public's inability to "exercise choice" by rejecting foods with any trace
of soy derived from these politically-incorrect
soybeans (Br. Med. J. 316: 1845, 1998). The lone article that
sparked all the street theater over monarch butterflies (Nature 399:
214, 1999) fed biotech pollen to their caterpillars under lab conditions;
decide for yourself. "Nature" itself called it a bad paper
(Nat. Biot. 17: 627 & 1154, 1999),
Science 290: 2088, 2000 called it a crock,
and the one group that tried to repeat the results under
field conditions found no sign of toxicity
(Proc. Nat. Acad. Sci. 97: 7700, 2000).
Of course, people who really care about butterflies
might have more to say about the massive destruction of the monarchs'
winter habitats in Mexico.
Look for much more interesting applications of gene therapy
(Lancet 338: 1427, 1991; Br.
Med. J. 303: 1282, 1991), perhaps modifying our own genetic expression during our later years. It
would be nice if Alzheimer's brain changes were no longer an inevitable part of getting old.
As a leader of the 21st century, you will need to think soberly about genetic disease and the very special
problems it raises. Reasonable people will differ (some) about morals and ethics. But not everybody
is reasonable, or even honest. If you offer your time, money, or prestige to any group of activists,
please be sure you clearly understand the group's real purposes, not just what they say in their
rhetoric; in today's world, the reality is likely to be quite different. I think we all hope that, when
difficult decisions must be made, that they will be well-informed. Society will increasingly look to you,
the physician, for this knowledge. Know your stuff yourself.
 Chromosomes/ translocations
Chromosomes/ translocations
"Pathology Outlines"
Nat Pernick MD
There are now five markers for Down's, all usable during the first
trimester (α-fetoprotein, pregnancy-associated
protein A, hCG, free beta chain of hCG, and unconjugated estriol);
levels tell risk: NEJM 338:955, 1998.
{53766} Down's child
{53767} Down's child
{13442} Brushfield's spots
{13443} palmar crease
{20093} Down's, hole in heart (ventricle and atrium have been opened to reveal AV-cushion defect)
{31573} Down's syndrome, narrow superior temporal gyrus (subtle, subtle)
{13448} Down's karyotype
 "Simian crease"
"Simian crease"
WebPath Case of the Week
* These children usually simply stop breathing shortly
after birth. Activists' attempts to keep these children alive
(Arch. Dis. Child. 75: F38, 1996) seem particularly cruel
and futile.
{13451} trisomy 18, hand with overlapping fingers
{13467} trisomy 18, newborn
{13457} trisomy 18; child made it to age 2; trust me
{20101} trisomy 18; hole in heart
{53772} trisomy 18; omphalocele (abdominal wall didn't form properly around umbilicus)
{13465} trisomy 18 karyotype
Trisomy 18
WebPath Photo
{13420} Trisomy 13
{16617} Trisomy 13, polydactyly
{16619} Trisomy 13, microcephaly
{20099} Trisomy 13, hole in heart
{13423} Trisomy 13 karyotype
 Partial Trisomy 13
Partial Trisomy 13
Rebecca's mother is a cyberfriend.
Recommended.
{16605} Trisomy 9. Both for your interest. What do you see?
 Full Metal Jacket
Full Metal Jacket
Jelly Doughnut Scene
{13406} Cornelia de Lange syndrome
{13405} Cornelia de Lange syndrome
* These kids don't make great
athletes; they are wiry rather than bulky, and tend to be poorly coordinated.
Pectus, squint, and elbows turned a bit farther out than most other guys
are supposedly common features as well.
* Richard Speck, the vile murderer of eight student nurses,
was tall and had
acne; he defended himself at trial saying he was an XYY, which he
actually was not. This contributed to the "guy with the most acne
on the prison basketball team" stereotype; of course the XYY defense
-- even if the perpetrator has it --
is long-discredited (Ciba Foundation Symposium 194: 248, 1996).
You can find plenty of accounts of
individual kids with developmental delay / behavior
problems who turn out to be XYY's. And you'll find "series" of patients
in which most of the XYY's have behavior problems. This is a classic
example of drawing faulty conclusions from a selected patient population.
Until somebody shows that kids who are karyotyped
as part of a fishing expedition to explain behavior problems are MUCH
more likely than the 1-in-a-thousand to turn out to be XYY, I conclude
that the relationship is dubious at best. Indeed, the largest study
indicates that the frequency is the same as in the general population --
indicating XYY is NOT a measurable risk factor for mental retardation
or major character problems (Genetic Counseling 6: 197, 1995).
If it's a minor risk factor for minor problems (as suggested
in the big Danish study in Birth Defects 26: 209, 1990) --
who cares?
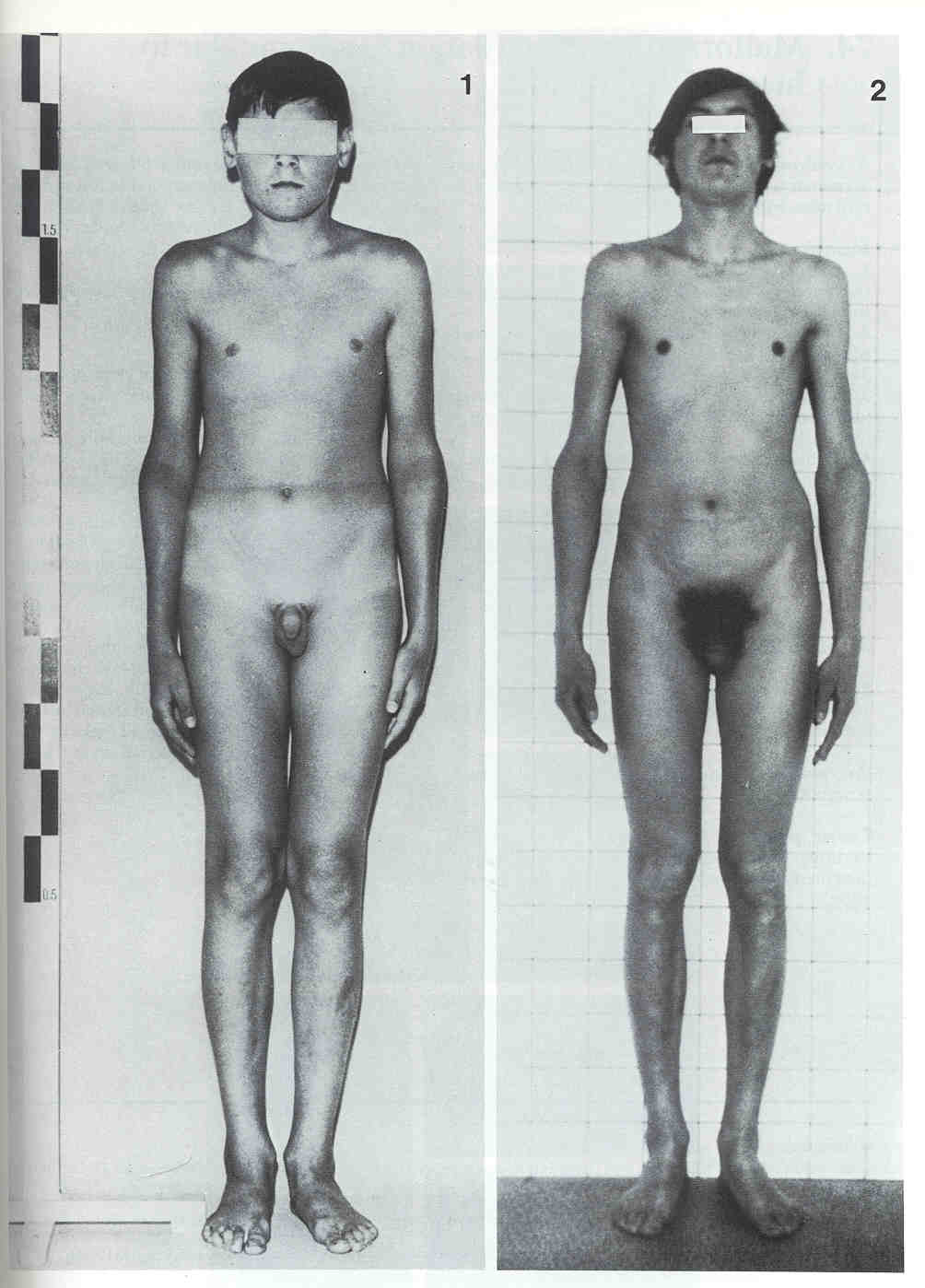 XYY
XYY
6'2" at age 14; 6'7" at age 18
From Wiedemann's classic textbook
{13435} Turner's; short fourth
{20105} Turner's, small uterus flanked by two streak ovaries
{20106} Turner's; streak ovary histology (right, no eggs), normal for comparison (left, eggs)
* Life birth of a non-mosaic tetraploid child: Arch. Path. Lab. Med. 127:
1612, 2003.
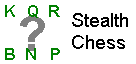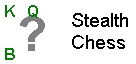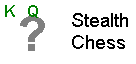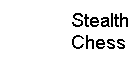 Ductal sex is determined by whether the muellerian (male)
or wolffian (female) ducts developed. (* Remember
that the embryonic testis's Sertoli cells produces "muellerian regression factor").
Ductal sex is determined by whether the muellerian (male)
or wolffian (female) ducts developed. (* Remember
that the embryonic testis's Sertoli cells produces "muellerian regression factor").
{13464} Marfan's, spine with scoliosis
{39124} Marfan guy
{40362} Marfan fingers

* Animal model for defective LDL receptors:
the Watanabe rabbit (Science 256: 772, 1992.)
{53675} albino
{31973} Niemann-Pick's foam cells
{17427} Niemann-Pick's, lysosomal storage in marrow (Giemsa stain and electron micrograph)
 Niemann-Pick's
Niemann-Pick's
Virginia Pathology Cases
The most common lysosomal storage disease.
{12224} Gaucher cell, bone marrow
{13631} Gaucher's, bone marrow biopsy, H&E (the lone
cell with the large hyperchromatic nucleus is a megakaryocyte)
{16216} Gaucher's cell, watered-silk appearance
{31979} Gaucher cell
{31980} Gaucher cell
{18244} Gaucher's child, large spleen and liver make abdomen protrude
The mucopolysaccharidoses

Ian Michael Smith (left)
Actor with Morquio's
{11539} Von Gierke's disease involving kidney, histologic view; clear glycogen in proximal tubular
epithelium
{20132} Von Gierke's disease, intracytoplasmic glycogen appears clear
{18238} Von Gierke's patient with hepatomegaly producing protuberant abdomen
I-cell disease ("mucolipidosis II")
is among the most popular biochemistry exam items. It is a storage disease
in which several different enzymes are lacking in lysosomes.
Along with its milder allele "Pseudo-Hurler", it results from lack of
N-acetylglucosamine phosphotransferase. This is the enzyme responsible
for picking up the mannose-6-phosphate-tagged enzymes that are destined to go into the lysosomes.
* Replacement enzyme therapy: Lancet 358: 601, 2001.
Fabry's
Pittsburgh Pathology Cases
Fabry's
Electron micrographs
VCU Pathology
{53765} fragile-X guy
* The mitochondrial genome follows the old routes
of trafficking in female slaves: Am. J. Hum. Genet. 72: 1058, 2003.
Mitochondrial myopathy
Wash. U., St. Louis
Illustrated notes


| Visitors to www.pathguy.com reset Jan. 30, 2005: |
Ed says, "This world would be a sorry place if
people like me who call ourselves Christians
didn't try to act as good as
other
good people
."
Prayer Request
 Teaching Pathology
Teaching Pathology
PathMax -- Shawn E. Cowper MD's
pathology education links
Ed's Autopsy Page
Notes for Good Lecturers
Small Group Teaching
Socratic
Teaching
Preventing "F"'s
Classroom Control
"I Hate Histology!"
Ed's Physiology Challenge
Pathology Identification
Keys ("Kansas City Field Guide to Pathology")
Ed's Basic Science
Trivia Quiz -- have a chuckle!
Rudolf
Virchow on Pathology Education -- humor
Curriculum Position Paper -- humor
The Pathology Blues
 Ed's Pathology Review for USMLE I
Ed's Pathology Review for USMLE I
![]()
![]()
 | Pathological Chess |
 |
Taser Video 83.4 MB 7:26 min |

 Debra Collins
Debra Collins
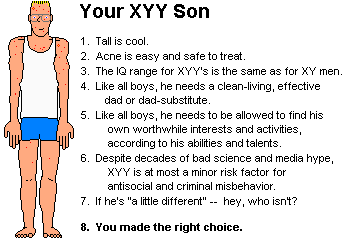
 Achondroplastic dwarf
Achondroplastic dwarf Ehlers-Danlos
Ehlers-Danlos Gaucher's Disease
Gaucher's Disease
 NIH Gene List
NIH Gene List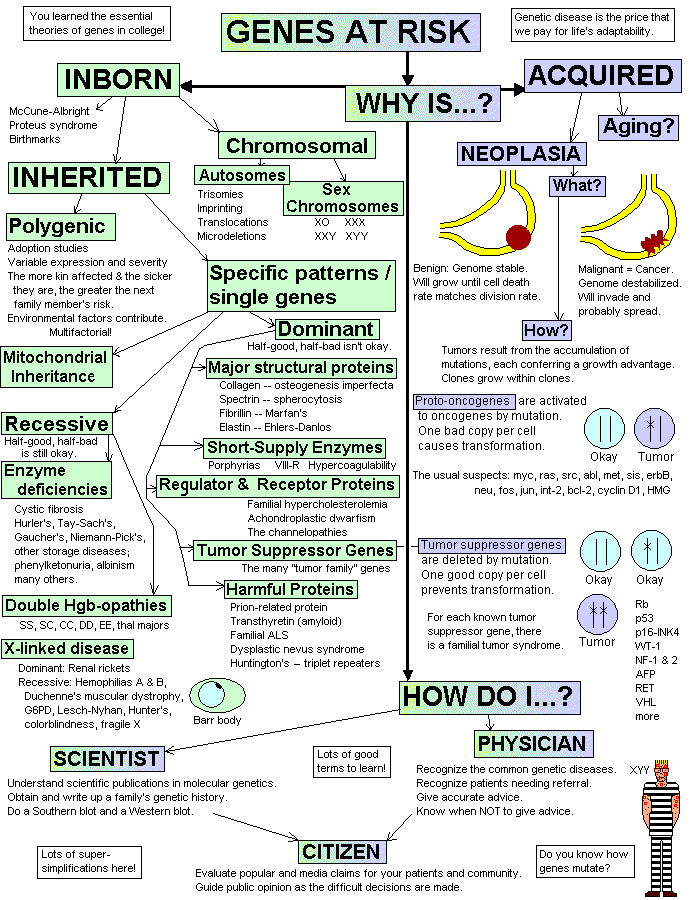 Genes at Risk
Genes at Risk