(EXCEPT AIDS)
Ed Friedlander, M.D., Pathologist
scalpel_blade@yahoo.com



Cyberfriends: The help you're looking for is probably here.
Welcome to Ed's Pathology Notes, placed here originally for the convenience of medical students at my school. You need to check the accuracy of any information, from any source, against other credible sources. I cannot diagnose or treat over the web, I cannot comment on the health care you have already received, and these notes cannot substitute for your own doctor's care. I am good at helping people find resources and answers. If you need me, send me an E-mail at scalpel_blade@yahoo.com Your confidentiality is completely respected.
 DoctorGeorge.com is a larger, full-time service.
There is also a fee site at myphysicians.com,
and another at www.afraidtoask.com.
DoctorGeorge.com is a larger, full-time service.
There is also a fee site at myphysicians.com,
and another at www.afraidtoask.com.
Translate this page automatically
 |
With one of four large boxes of "Pathguy" replies. |
 I'm still doing my best to answer
everybody.
Sometimes I get backlogged,
sometimes my E-mail crashes, and sometimes my
literature search software crashes. If you've not heard
from me in a week, post me again. I send my most
challenging questions to the medical student pathology
interest group, minus the name, but with your E-mail
where you can receive a reply.
I'm still doing my best to answer
everybody.
Sometimes I get backlogged,
sometimes my E-mail crashes, and sometimes my
literature search software crashes. If you've not heard
from me in a week, post me again. I send my most
challenging questions to the medical student pathology
interest group, minus the name, but with your E-mail
where you can receive a reply.
Numbers in {curly braces} are from the magnificent Slice of Life videodisk. No medical student should be without access to this wonderful resource. Someday you may be able to access these pictures directly from this page.
Also:
Medmark Pathology -- massive listing of pathology sites
Freely have you received, freely give. -- Matthew 10:8. My
site receives an enormous amount of traffic, and I'm
handling about 200 requests for information weekly, all
as a public service.
Pathology's modern founder,
Rudolf
Virchow M.D., left a legacy
of realism and social conscience for the discipline. I am
a mainstream Christian, a man of science, and a proponent of
common sense and common kindness. I am an outspoken enemy
of all the make-believe and bunk that interfere with
peoples' health, reasonable freedom, and happiness. I
talk and write straight, and without apology.
Throughout these notes, I am speaking only
for myself, and not for any employer, organization,
or associate.
Special thanks to my friend and colleague,
Charles Wheeler M.D.,
pathologist and former Kansas City mayor. Thanks also
to the real Patch
Adams M.D., who wrote me encouragement when we were both
beginning our unusual medical careers.
If you're a private individual who's
enjoyed this site, and want to say, "Thank you, Ed!", then
what I'd like best is a contribution to the Episcopalian home for
abandoned, neglected, and abused kids in Nevada:
My home page
Especially if you're looking for
information on a disease with a name
that you know, here are a couple of
great places for you to go right now
and use Medline, which will
allow you to find every relevant
current scientific publication.
You owe it to yourself to learn to
use this invaluable internet resource.
Not only will you find some information
immediately, but you'll have references
to journal articles that you can obtain
by interlibrary loan, plus the names of
the world's foremost experts and their
institutions.
Alternative (complementary) medicine has made real progress since my
generally-unfavorable 1983 review linked below. If you are
interested in complementary medicine, then I would urge you
to visit my new
Alternative Medicine page.
If you are looking for something on complementary
medicine, please go first to
the American
Association of Naturopathic Physicians.
And for your enjoyment... here are some of my old pathology
exams
for medical school undergraduates.
I cannot examine every claim that my correspondents
share with me. Sometimes the independent thinkers
prove to be correct, and paradigms shift as a result.
You also know that extraordinary claims require
extraordinary evidence. When a discovery proves to
square with the observable world, scientists make
reputations by confirming it, and corporations
are soon making profits from it. When a
decades-old claim by a "persecuted genius"
finds no acceptance from mainstream science,
it probably failed some basic experimental tests designed
to eliminate self-deception. If you ask me about
something like this, I will simply invite you to
do some tests yourself, perhaps as a high-school
science project. Who knows? Perhaps
it'll be you who makes the next great discovery!
Our world is full of people who have found peace, fulfillment, and friendship
by suspending their own reasoning and
simply accepting a single authority that seems wise and good.
I've learned that they leave the movements when, and only when, they
discover they have been maliciously deceived.
In the meantime, nothing that I can say or do will
convince such people that I am a decent human being. I no longer
answer my crank mail.
This site is my hobby, and I presently have no sponsor.
This page was last updated February 6, 2006.
During the ten years my site has been online, it's proved to be
one of the most popular of all internet sites for undergraduate
physician and allied-health education. It is so well-known
that I'm not worried about borrowers.
I never refuse requests from colleagues for permission to
adapt or duplicate it for their own courses... and many do.
So, fellow-teachers,
help yourselves. Don't sell it for a profit, don't use it for a bad purpose,
and at some time in your course, mention me as author and KCUMB as my institution. Drop me a note about
your successes. And special
thanks to everyone who's helped and encouraged me, and especially the
people at KCUMB
for making it possible, and my teaching assistants over the years.
Whatever you're looking for on the web, I hope you find it,
here or elsewhere. Health and friendship!
 I am presently adding clickable links to
images in these notes. Let me know about good online
sources in addition to these:
I am presently adding clickable links to
images in these notes. Let me know about good online
sources in addition to these:
Pathology Education Instructional Resource -- U. of Alabama; includes a digital library
Houston Pathology -- loads of great pictures for student doctors
Pathopic -- Swiss site; great resource for the truly hard-core
Syracuse -- pathology cases
Walter Reed -- surgical cases
Alabama's Interactive Pathology Lab
"Companion to Big Robbins" -- very little here yet
Alberta
Pathology Images --hard-core!
Cornell
Image Collection -- great site
Bristol Biomedical
Image Archive
EMBBS Clinical
Photo Library
Chilean Image Bank -- General Pathology -- en Español
Chilean Image Bank -- Systemic Pathology -- en Español
Connecticut
Virtual Pathology Museum
Australian
Interactive Pathology Museum
Semmelweis U.,
Budapest -- enormous pathology photo collection
Iowa Skin
Pathology
Loyola
Dermatology
History of Medicine -- National Library of Medicine
KU
Pathology Home
Page -- friends of mine
The Medical Algorithms Project -- not so much pathology, but worth a visit
National Museum of Health & Medicine -- Armed Forces Institute of Pathology
Telmeds -- brilliant site by the medical students of Panama (Spanish language)
U of
Iowa Dermatology Images
U Wash
Cytogenetics Image Gallery
Urbana
Atlas of Pathology -- great site
Visible
Human Project at NLM
WebPath:
Internet Pathology
Laboratory -- great site My team:
My team:Ed Lulo's Pathology Gallery
Bryan Lee's Pathology Museum
Dino Laporte: Pathology Museum
Tom Demark: Pathology Museum
Dan Hammoudi's Site
Claude Roofian's Site
Pathology Handout -- Korean student-generated site; I am pleased to permit their use of my cartoons
Estimating the Time of Death -- computer program right on a webpage
Pathology Field Guide -- recognizing anatomic lesions, no pictures

St.
Jude's Ranch for Children
I've spent time there and they are good. Write "Thanks
Ed" on your check.
PO Box 60100
Boulder City, NV 89006--0100
More of my notes
My medical students
Clinical
Queries -- PubMed from the National Institutes of Health.
Take your questions here first.
HealthWorld
Yahoo! Medline lists other sites that may work well for you

We comply with the
HONcode standard for health trust worthy
information:
verify
here. 


![]()
Describe the typical clinical pictures, gross and microscopic pathology, immunopathology, and pathophysiology of each of the following:
polyarteritis nodosa (periarteritis nodosa)
Wegener's ("pathergic") granulomatosis
Explain the importance of making an accurate diagnosis of each of the two diseases listed above, and tell how to go about doing it.
Define amyloid and give its composition and physical chemistry. Describe its light and electron microscopic appearances. Tell where to find it and how to recognize it. Recognize "primary amyloidosis" and "secondary amyloidosis" as archaic terms.
Given an amyloid derived from each of the following, tell its names, and describe the associated syndrome in some detail.
immunoglobulin light chains
SSA protein
transthyretin ("prealbumin")
hormone polypeptide
beta2 microglobulin
Describe where amyloid is deposited in each of these organs, and its effect on function:
kidney
heart
blood vessels
tongue
gut
liver
adrenals
spleen
vessels
skin
nerves
flexor retinaculum
Recognize possible amyloidosis patients. Tell how to make the diagnosis of systemic amyloidosis. Tell how to treat them.
Give the distinguishing features of each of the following hereditary immunodeficiencies:
Recognize and describe common variable immunodeficiency.
Mention the distinguishing features of the following hereditary deficiencies of the complement system:
Recognize each of the following using the microscope:
POLYARTERITIS NODOSA ("periarteritis nodosa")
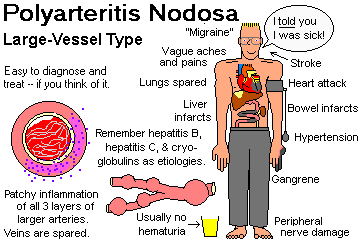
A serious disease characterized by focal, three-layer, necrotizing inflammation, with deposition of immune complexes and complement, involving medium-sized and small arteries in many different organs.
This inflammation closely resembles the classic type III immune injury.
{16917} fibrinoid necrosis of artery (polyarteritis nodosa)
{53545} fibrinoid necrosis of artery (polyarteritis nodosa)
Indeed, around a third of "classic" acute cases were caused by deposition of hepatitis B surface antigen and antibody in the walls of vessels. It still happens a lot but responds nicely to common-sense treatment (Medicine 74: 238, 1995).
Patients may be of any age. There is a slight male preponderance. The symptoms depend on what little arteries are involved.
The disease involves only short portions of affected arteries. We can conjecture that a small intimal injury begins a chain reaction, with more polys going to the involved areas and making things worse. And obviously if the whole arterial system were involved, death would already have occurred.
Damage to the vessel walls results in thrombosis and infarctions.
The most common site of involvement is the kidney (pain, blood in the urine, high blood pressure).
Destruction of collagen and elastin in the walls of the damaged vessels causes the "nodose" ballooning ("microaneurysms").
{16920} polyarteritis, angiogram showing aneurysms
The untreated disease is highly lethal and the diagnosis is often missed. Death results from stroke, high blood pressure, kidney failure, or massive bleeding from GI infarcts.
Classic polyarteritis -- "in the differential diagnosis of everything" -- is still a diagnosis of exclusion.
Unfortunately the disease simulates "aches and pain of the 'flu" (not surprising, since there is a vasculitis), and goes neglected for weeks or even years.
One feature that distinguishes polyarteritis nodosa from other inflammatory diseases of the vessels is that it does not involve the lungs.
{11987} polyarteritis nodosa, infarcts on
the elbows
{11988} polyarteritis, gangrene
Despite the value of ANCA in spotting polyarteritis nodosa, we still suggest a biopsy. The best place to biopsy is probably a tender muscle or testis -- or do a renal angiogram and see the nodules on the arteries.
Microscopically, look for lots polys, eos, and/or monos in all three layers of the arterial wall. Leakage of protein from the plasma into the damaged vessel produces "fibrinoid" ("fibrinoid necrosis", etc.,) typical of any type III immune injury. However, there's usually not much immunoglobulin, so perhaps this isn't really the mechanism.
After the lesions heal, you can still see the damaged elastic membranes. Beware! There may well be active lesions elsewhere in the body!
{46396} old polyarteritis nodosa, burned-out scarred lesion showing disrupted elastica (elastic stain,
of course)
{16918} old polyarteritis nodosa, elastic stain
{16922} old polyarteritis nodosa, elastic stain (the
old break in the internal elastic membrane is obvious;
organized thrombus in the center stains with elastic stain also)
It is important to make the diagnosis, because this deadly disease responds very well to treatment (* prednisone plus cyclophosphamide).
*The equally-deadly Small vessel polyarteritis (anti-myeloperoxidase disease, p-ANCA disease, microscopic polyangiitis) will be mentioned under Wegener's granulomatosis; the involved vessels are smaller, little veins can be involved as well as arteries, the lung is often involved (lung pathology Arch. Path. Lab. Med. 121: 144, 1997 and almost all the patients have p-ANCA.

* Kawasaki disease ("mucocutaneous lymph node syndrome") of children has identical histology to polyarteritis nodosa, but we will cover it later (NEJM 315: 1143, 1986, classic article).
*It also would be logical to talk about Henoch-Schonlein purpura here. This is a fairly common, mild illness caused by IgA deposition in specific sites. More about this later.
WEGENER'S GRANULOMATOSIS ("pathergic granulomatosis" -- JAMA 273: 1288, 1995; Am. J. Med. Sci. 321: 76, 2001)

This is a multi-system vasculitis caused (somehow) by antibodies against proteinase 3 (* "p29"; apparently the same as "myeloblastin" 88: 9253, 1991; review Am. J. Path. 139: 831, 1991). Exactly why this produces granulomas is especially puzzling.
Reports that differ markedly from these tend to come from obscure institutions. Nowadays the literature suggests that you want a biopsy even if it looks like Wegener's clinically and there's a positive c-ANCA (Lancet 346: 926, 1995). Reviews with caveats: J. Clin. Path. 52: 124, 1999, Pathology 31: 38, 1999; Am. J. Clin. Path. 111: 507, 1999 (consensus statement).
You can also get an ELISA anti-PR3 tube test; it's sensitive for Wegener's, but not very specific: Postgrad. Med. J. 76: 287, 2000.
Wegener's is characterized by acute necrosis and inflammation (* all cell types) of short portions of the walls of arteries and veins, primarily involving the respiratory tract, kidneys, and * spleen.
In addition, there are extravascular, necrotizing granulomas in the respiratory tree, especially the nasal sinuses and lung parenchyma. These tend to ulcerate / cavitate. (* One can mistake it for TB or fungus infection -- to make the diagnosis of Wegener's, there must be no demonstrable micro-organisms.)
These patients commonly die from exsanguinating pulmonary hemorrhage.
*A horrid, rare "disease" that is "related to Wegener's" is lethal midline granuloma in which the nose, then the center of the face, sloughs. Recently it's become clear this is usually a T-cell lymphoma, inciting a granulomatous response -- and not a Wegener's variant at all. A few cases remain "idiopathic" -- and these are probably real Wegener's variants (Ann. Plast. Surg. 37: 532, 1996).
Most (not all) cases show severe injury to the renal glomeruli (* segmental-necrotizing and/or granulomatous and/or rapidly-progressive glomerulonephritis).
A majority of patients have involvement of their eyes, and many have problems with their ears (pinna, Eustachian tube problems, nerve VIII). The peripheral nerves are sometimes damaged (by microinfarcts). Several cases with skin involvement have been mistaken for pustular acne.
* Even without a positive c-ANCA blood test, today's savvy pathologist calls "Wegener's" if he/she sees vasculitis, necrosis, and granulomas in the right mix and setting. If only two are present, the call can be made if there is lung, head-and-neck, and kidney involvement; otherwise it'll be "probable" Wegener's. If only one is present, the biopsy can at most be "suggestive".
{40524} Wegener's; vasculitis lesion is in center
{40520} Wegener's; giant cells and damaged tissue
{40525} Wegener's; burned-out lesion (elastic stain, showing disrupted elastica)
{40526} Wegener's, vasculitis, note giant cell at top of picture
{40527} Wegener's; necrosis and giant cell
{11990} Wegener's, necrosis of elbow
The molecular biology is now worked out. The anti-proteinase 3 autoantibody occurs in almost all
cases,
and titers rise and fall along with disease activity.
Neutrophils express some proteinase 3 on their surfaces, and are probably attacked and eviscerated by the
antibodies. They spill their granules, which would be unwholesome. The body sites in which the
most neutrophils ordinarily occur are the nose and the lungs; glomerular involvement may result
from trapping immune complexes.
Apparently, endothelial cells also express proteinase 3 on their surfaces in response to various
cytokines, and this must be part of the problem, too (Blood 82: 1221, 1993'
Clin. Exp. Imm. 115: 362, 1999; presently it's much-discussed whether
this is endogenous or exogenous; the latter makes more sense Clin. Imm. 97: 171, 2000).
* Some macrophages also express proteinase 3 on their surfaces, perhaps accounting for the
granulomas Am. J. Path. 139: 723, 1991.
Patients are usually young adults. They complain first of stuffy nose and other ill-defined (or
"hypochondriac") symptoms. Because inflammation of the vessels is prominent, the disease simulates "aches
and pains of the 'flu", and goes unrecognized.
The extent of disease is conventionally described using the mnemonic ELK: Ears-nose-throat,
Lungs, Kidneys.
Although serology is helpful, we still suggest a biopsy.
The most important thing to remember about "Wegener's" is that it is treatable using
cyclophosphamide (* with or without prednisone), and you must make the diagnosis.
Tissue diagnosis is usually accomplished by open biopsy. The pathologist
will want to find two of these three:
* This is preferable to
renal biopsy -- the renal lesions are usually impossible to tell from polyarteritis nodosa or
Churg-Strauss, and the kidneys may be spared completely. You may try a transbronchial biopsy
first, though the yield is low.
In the microvascular version of polyangiitis nodosa (now widely considered
"like Wegener's, but
is tougher on the kidneys"),
they are usually directed against myeloperoxidase
p-ANCA, p-ACPA, so-named because of
artifactual perinuclear distribution of the antigen on air-dried slides) and/or elastase (Thorax 46: 70,
1991, many others).
Current thinking is that, in both diseases, cytokines ("released because of some co-existing
infection") prime neutrophils to begin expressing these auto-antigens on their surfaces. They are
then made to degranulate by autoantibodies reacting with their surfaces.
*Antibiotics as an effective treatment for Wegener's:
Arch. Int. Med. 151: 1649, 1991, many others.
Perhaps a soluble bacterial antigen is the cause after all.
Of course, etanercept is under study (Arth. Rheum. 44: 1149, 2001).
* The Churg-Strauss phenomenon is
a vascuilitis with a preponderance
of eosinophils. ANCA is usually positive.
The popular wisdom that this is simply Wegener's or polyarteritis in people
with atopy/asthma
(Thorax 46: 70, 1991) does not fit with the fact that the asthma
is usually new (and permanent: Lancet 361: 587, 2003), and the disease responds
better than its eosinophil-poor
counterparts to glucocorticoids.
There are numerous eosinophils in the inflammatory lesions. Lung involvement is common, and
small veins are involved as well as small arteries. Thankfully, almost everybody
survives the illness nowadays. If leukotriene receptor antagonists are ever
a cause, it is rare (Am. J. Med. 115: 284, 2003). Pathologists review
Am. J. Clin. Path. 114: 767, 2000. Clinicians: Am. J. Med. 115: 284, 2003.
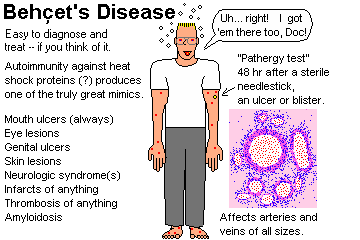
BEHÇET'S DISEASE (Behcet's disease; NEJM 341: 1284, 1999)
It's probably due to local production
of T-cells angry with against heat shock proteins (HSP-60)
(Med. Exp. Imm. 113: 100, 1998. others).
Streptococci seem to be triggers somehow, so some of these
people get maintained on penicillin.
Pathology: Arth. Rheum. 39: 1926, 1996.
Criteria:
Must have two of these:
*There's a strong link to HLA-B51. It's a major cause
of blindness in Japan. Behçet's can do lots of things, including
producing a host of neurologic problems: Arch. Neuro. 53:
691, 1996, and/or thrombosis of anything.
Interferon-α for Behçet's syndrome:
Arch. Derm. 134: 1010, 1998. Thalidomide: Ann. Int. Med.
128: 443, 1998; Lancet 363: 1802, 2004. Cyclosporine Br. J. Rheum. 36:
1113, 1997. Azathioprine: Arth. Rheum. 40: 769, 1997.
 Wegener's
Wegener's
Pittsburgh Pathology Cases
Wegener's
Lung pathology series
Dr. Warnock's Collection
Churg-Strauss
Lung pathology series
Dr. Warnock's Collection
A poorly-understood, insidious vasculitis syndrome featuring some or all
of the following:
Must have oral ulcers (* watch for newer definitions that allow for the rare case without oral ulcers)
Non-substituted: Old hearts ("senile cardiac amyloidosis")
There are many, many other rare familial amyloidosis syndromes
(Blood 90:4799, 1997).
Reviews: Postgrad. Med. 96: 119, 1994; Mayo Clin. Proc. 74: 490, 1999.
Molecules NEJM 349: 583, 2003.
Amyloid is a class of insoluble, homogeneous, eosinophilic ("hyaline") substances that accumulate
in extracellular spaces. Their common link is that they are beta-pleated sheets (rather than α
helices), and therefore cannot be effectively handled by the body.
* All amyloids are now known to be built up from soluble oligomers
with a particular conformation (Science 300: 486, 2003) that seems
necessary to their becoming beta-pleated.
Amyloids don't go away, and eventually the surrounding cells undergo "pressure atrophy".
Whatever is really happening, enough amyloid in an organ makes it rubbery and waxy and
(sometimes) useless.
*There is much interest right now in how amyloid is laid down, and
you'll read about a substance called "amyloid enhancing factor" that must
be produced first (Arthr. Rheum. 48: 1430, 2003, lots more).
Amyloidosis is a group of curious systemic and (?) localized diseases with varied and bizarre
clinical presentations.
The classic forms of amyloidosis are not among the commonest diseases, though they are not rare.
Alzheimer's disease, now recognized to be one of the commonest and deadliest diseases, regularly
includes amyloid beta/A4 deposition in the brain and its vessels. (It's real amyloid, Am. J. Path.
143: 1594, 1993).
Amyloidosis involving the heart is responsible for an unknown number of cases of heart disease in
the elderly.
Amyloid was named by Dr. Virchow, who noticed that iodine turned it orange-brown and
subsequent application of sulfuric acid turned it purple-blue. Because starch undergoes the same
color reactions, amyloid means "starch-like".
*Amyloidosis as a disease was discovered by Dr. Rokitansky, who called it "lardaceous change".
The name "amyloidosis" is inappropriate today, because amyloid and starch are different
chemically. But nobody liked the suggestion "beta-fibrillosis".
Most amyloids are altered forms of various proteins that exist in the healthy body.
Regardless of its origin and amino-acid sequence, amyloid (and, in us chordates, only amyloid and
perhaps tooth enamel) is not an alpha-helix, but a crossed beta-pleated sheet. (Remember Dr. Linus
Pauling's two stable secondary structures for proteins?)
This beta-pleated structure results in the marked affinity all amyloids exhibit for Congo Red dye,
and for the apple-green birefringence it exhibits when so stained and examined under polarized light.
{08129} Congo red
*Amyloid stains metachromatically (i.e., a different color from the dye solution, in this case red
rather than violet) with crystal violet, and has other exotic reactions with special stains (notably
fluorescence with thioflavin T). Ask a pathologist if you're curious.
{17480} crystal violet stain, amyloid (red) in heart
Despite its amorphous appearance on light microscopy, electron microscopy shows that 90% of any
type of amyloid consists of non-branching fibrils, 70 to 100 angstroms across. These are usually
crisscross but may be parallel.
These fibrils are the beta-pleated protein, the congophilic component of amyloid.
The remaining 10% of any amyloid is composed of "P-component", a 90-angstrom, pentagonal
donut-rod thing.
The P component is pentamers of a glycosylated α-1 globulin ("serum amyloid P-component",
SAP), an acute-phase protein similar in sequence to C-reactive protein that is produced by the liver in response
to interleukin 1 production by macrophages. It imparts the weak
PAS-positivity to amyloid.
All about P-component:
Proc. Nat. Acad. Sci. 91: 5602, 1994.
The P component, bound to the fibrils,
is also required to prevent the amyloid
from being degraded by the body. (Ciba F.S. 199: 73, 1996).
* Surprise! P's job in health seems to be to cover over the nuclear dust released
from dead cells and control the breakdown of the chromatin, preventing
antinuclear antibodies from forming.
Nat. Med. 5: 694, 1999.
Maybe this is why amyloidosis
almost never occurs in lupus (Ed's idea).
As noted, all types of amyloid look the same at any magnification.
{08159} iodine treatment
has blackened amyloid in glomeruli
Amyloidosis often goes unrecognized.
The typical patient with one of the classic forms of amyloidosis takes about a year to be correctly
diagnosed.
Isolated cardiac amyloidosis is the problem in an unknown number of elderly patients with heart
disease, especially heart block, due to "old age". Biopsy is not usually attempted on these patients.
Alzheimer's disease is ultimately an autopsy diagnosis, and amyloid is a regular finding in these
patients' brains (see below).
Classification
*The old categories of "primary" and "secondary amyloidosis" are archaic.
"Primary amyloidosis" was "idiopathic" or cased by cancers of B-cell origin. "Secondary
amyloidosis" arose in patients with chronic infections.
"Secondary amyloidosis" supposedly involved liver, spleen, adrenals, and kidneys, while "primary
amyloidosis" involved these, the heart, tongue, gut, muscles, skin, nerve, ligaments, and other
organs. Even this is not reliable.
Today, amyloids are classified instead by the protein from which they are derived.
Amyloid derived from immunoglobulin light-chains ("amyloid B", "amyloid AL",
"immunocyte-derived amyloidosis", "B-cell dyscrasia with amyloidosis", the old "primary
amyloidosis", etc.).
This is likely to involve many different organs. The amyloid is composed of some or all of the
lambda or (*less often) kappa light chains of immunoglobulin molecules.
"Amyloid, light-chain derived" is called "amyloid AL" or "amyloid B". (* Not all diseases with
light-chain deposition are amyloidosis.
Most patients have evidence of a hyperactive clone of B-cells.
Many have overt plasma-cell myeloma producing light chains (around 20% of plasma cell myeloma
patients eventually get amyloidosis), or related diseases such as B-cell lymphomas or
* Waldenstrom's macroglobulinemia.
Even more have a "benign monoclonal gammopathy", and this includes most patients who have
"idiopathic primary amyloidosis".
Nowadays, it's probably good practice to rule out the hereditary
amyloidosis syndromes (transthyretin, fibrinogen, lysozyme, apo-A1)
before calling anything "light-chain amyloidosis": NEJM 346:
1786, 2002.
Amyloidosis derived from AAP protein ("amyloid A", "amyloid AA", "reactive systemic
amyloidosis", the old "secondary amyloidosis", * "permanganate sensitive amyloidosis", etc.; old review
Medicine 70: 246, 1991)
The amyloid is composed of a portion of "serum amyloid associated protein" (SAA ; AAP; a serpin).
This large serum protein ordinarily is present in very low quantities. The liver increases AAP
production by several orders of magnitude during the "acute phase reaction" in response to
interleukin 1 from macrophages.
*Macrophages release interleukin 1 when they are phagocytizing something, and it alters the levels
of most of the proteins in the plasma. More about this when we see you again.
*Of the three common amyloids, this is the only one that loses congophilia on treatment with
potassium permanganate.
Most of the patients with classic "secondary" amyloidosis have this type of amyloid.
These patients have some disease with ongoing breakdown of cells and thus longstanding activation
of their acute-phase proteins, including AAP. After a few years, amyloidosis may develop.
Important causes of reactive systemic amyloidosis are longstanding tuberculosis, leprosy, syphilis,
osteomyelitis, bronchiectasis, long-term cystic fibrosis survivors,
paraplegia (bedsores and bladder infections),
Crohn's enteritis (Gastroenterology 112:1362, 1997),
and
heroin skin-poppers.
More recently recognized causes of reactive systemic amyloidosis are rheumatoid arthritis (up to one
third of the really bad cases), familial
mediterranean fever,
* polymyositis, * inflammatory bowel disease, * renal cell carcinoma,
* Hodgkin's disease, * BehÇet's disease (Nephron 52: 154,
1989), and * chronic Reiter's syndrome (Am. J. Kid. Dis. 14: 319, 1989).
Obviously, not everyone with longstanding elevations of AAP gets amyloidosis A. Something else
must be wrong, probably with AAP processing by macrophages.
*Of course, serum AAP is not a suitable blood test for amyloidosis; it will be elevated in anyone
during the acute phase reaction.
*A genetic variant of AAP results in amyloid deposition in the thyroid gland ("amyloid goiter of
New Guinea"). Another familial variant causes one form of Ostertag amyloidosis (NEJM 317:
1520, 1987); other "Ostertag" amyloids result from mutant apo-A1 (no kidding, Proc. Nat. Acad.
Sci. 89: 7389, 1992) or lysozyme (Nature 362: 553, 1993; physics Nature 385: 787, 1997).
Suppressing amyloid formation and the damage that amyloid causes by blocking
the surface molecule on which it beta-pleats: Nat. Med. 6: 643, 2000.
Probably because it's antibodies rather than macrophages
doing the damage, lupus patients very rarely get amyloidosis.
Amyloid derived from transthyretin ("prealbumin";
including "amyloid AF", "amyloid ATTR", "familial amyloid",
"amyloidosis C", "old age amyloid", "senile amyloid" (Virchows Archiv. 442: 252, 2003), * "amyloid Sc1"=senile cardiac, first type)
Beta-pleated transthyretin (* "prealbumin") appears in many elderly people.
Though widespread, it is seldom significant except in heart, where it
is accounts for an unknown number of cases of unexplained heart block (failure to conduct the
impulse from atria to ventricles), other arrhythmias, and sudden death.
Massive deposits of "senile amyloid" interfere with ventricular function, but just a little can disrupt
the conduction system. If diagnosed (* echocardiography helps), it is called "senile cardiac
amyloidosis".
The problem is especially common in older black people, who often carry a mutation: NEJM 336:
466, 1997.
*Clinicians now talk about "senile systemic amyloidosis", noting that it is unlikely to cause non-cardiac problems. It is
usually missed clinically (Int. J. Cardiol. 32: 83, 1991). Old talk about this
all being the result of an amino acid substitution in transthyretin was wrong (Proc. Nat. Acad. Sci.
87: 2843, 1990).
A substituted amyloid C is a very common cause of heart block in older blacks.
New England Journal of Medicine 336:466, 1997.
Amyloid C also occurs in other clinical settings.
It is the amyloid of most familial amyloidosis syndromes ("amyloid AF"; as noted above, some have
amyloid A instead). A transthyretin with an amino-acid substitution (* typically valine --> leucine
in position 30) is deposited as amyloid. Polyneuropathy is the principal
feature of this type of amyloidosis, and of course these are autosomal dominant diseases.
(* Roundsmanship: Look for scalloped pupils in familial amyloidosis!)
*Some localized cerebral amyloidosis cases are amyloidosis C.
*New variants keep turning up. For example: "Appalachian Amyloidosis" -- Arthr. Rheum. 30:
195, 1987. Danish amyloidosis C of the heart: Am. J. Med. 93: 3, 1992. Another amyloidosis C of
the heart: Circulation 91: 962, 1995. Abyssinian cats get a familial amyloidosis. Molecular
biology of various amyloid C's: Clin. Immunol. Immunopathol. 39: 479, 1986.
*Liver transplantation as a highly effective therapy for familial amyloidosis C: Lancet 341: 1113,
1993.
Amyloid derived from a hormone polypeptide
Occasionally such material fills much of the stroma of tumors of the corresponding endocrine organ.
Carcinoma of the C-cells of the thyroid (* "medullary carcinoma of the thyroid") has a stroma rich
in beta-pleated precalcitonin.
The islets of many adult-onset diabetics have abundant amyloid
made from amylin ("islet-associated amyloid polypeptide, IAPP")
You may find it in the stroma of islet-cell tumors, too.
Amyloid composed of beta-2 microglobulin ("Amyloid H")
Amyloid arthropathy, mostly limited to the joints and flexor retinaculum, is a problem in
kidney-failure patients who have been on hemodialysis for years.
Beta-2 microglobulin is the light chains of HLA antigens. It is increased in the serum
of chronic renal failure, and it is the beta-pleated substance in cases of dialysis-related amyloid
arthropathy that have been studied.
Unlike most amyloids, this does seem to regress when the hemodialysis is replaced by
transplanted
kidneys: Lancet 338: 335, 1991.
* Amyloid H appears more on the serosa of the gut than on the
mucosa, accounting for the difficulty of diagnosis.
Journal of Clinical Pathology. 50(10):873-5, 1997 Oct.
Amyloid composed of "amyloid A4 protein" ("amyloid beta protein") (Am. J. Path. 142: 1449,
1993)
This is the amyloid in the senile plaques, neurofibrillary tangles, and cerebral vessels of Alzheimer's
disease and Down's syndrome.
*The accumulation of the protein in the cerebral blood vessels ("congophilic angiopathy") is not the
main problem in Alzheimer's, but the same protein accumulates in "Dutch congophilic
angiopathy", where it produces hemorrhages.
Amyloid composed of infectious prions (PrP)
The amyloid within the brain in Creutzfeldt-Jakob disease. More later.
* Amyloid derived from atrial natriuretic factor ("AANF")
This hormone becomes droplets of amyloid in the atria. It is present in most hearts from elderly
people, and probably has no significance. See Exp. Mol. Path. 52: 266, 1990; Bioch. Biophys. Res.
Comm. 148: 1087, 1987.
You may know atrial natriuretic factor better as "atriopeptin".
Another amyloid also accumulates here ("isolated atrial amyloid", "AIAA"). * One group
of pathologists suggests it underlies much of the "idiopathic atrial fibrillation"
seen in older people (Circ. 106: 2091, 2002).
Other amyloidosis syndromes
Amyloids of uncertain composition
"Congophilic angiopathy" of the vessels of the brain (as in Alzheimer's disease) also occurs in
dementia pugilistica, and other brain diseases, in which its structure awaits clarification.
*A severe form of cerebral congophilic angiopathy may occur alone and produce dementia and
multiple intracerebral hemorrhages (see Neurology 35: 625, 1985). At least some of these patients
have mutant gamma-trace alkaline microprotein ("cystatin C"; NEJM 311: 1547, 1984; Proc. Nat.
Acad. Sci. 91: 1416, 1994).
*Amyloids composed of beta-pleated apolipoprotein A turn up occasionally in families, and they are
quite common in the lung vessels of old dogs (Am. J. Path. 141: 1013, 1992).
In a few familial syndromes with generalized amyloidosis, the protein of origin has not been
identified.
Localized amyloidosis is diffuse infiltration of an organ, perhaps the result of some mutation in a
protein limited to that organ. The best-known examples are isolated involvement of the tongue,
and isolated laryngeal amyloidosis (J. Lar. 117: 647, 2003; laser rx).
Several hereditary corneal diseases feature amyloid.
{38620} amyloid, tongue
An "amyloidoma" (yes, another non-tumor ending in "-oma") is a single mass of amyloid with a
foreign-body reaction around it. These are poorly understood.
* CNS amyloid B amyloidomas:
Cancer 82:362, 1998.
{38899} amyloidoma, histology; note foreign-body reaction
Organ involvement in systemic amyloidosis syndromes
Kidney
Amyloid gets deposited in the glomeruli, the blood vessels, and around the tubules.
Usually the glomerular basement membrane becomes too leaky to proteins, and the patient gets the
"nephrotic syndrome".
If the patient with renal amyloidosis survives, eventually the amyloid plugs up the glomeruli and
that's the end of the kidneys.
Renal involvement is a major cause of death in amyloidosis.
{10880} renal amyloidosis, congo red
Heart and vessels
Amyloidosis C limited to the heart is described above, and
when cardiac biopsy became commonplace in the 1980's,
it was recognized as relatively frequent.
In any form of systemic amyloidosis, involvement of the heart may be severe or mild.
Amyloid deposition begins in the subendocardium. In relatively mild cases, tiny deposits of amyloid
occur on the atrial endocardium that resemble dewdrops. But more often, there's nothing at all
abnormal grossly.
*Almost all elderly people have traces of amyloids of unknown composition in their aortas,
independent of atherosclerosis, as well as
deposits of atrial natriuretic peptide (amyloid IAA). These are probably harmless.
Of course, many will also have some transthyretin-based amyloid, and most of these
people are probably asymptomatic.
The heart in severe cardiac amyloidosis, however, is stiff and heavy. This is the usual cause of
"restrictive cardiomyopathy" (stiff-heart disease).
Half of patients with amyloidosis B have pump failure or dangerous rhythm disturbances.
* You'll make the diagnosis of severe cardiac amyloidosis by biopsy
or echo.
{08111} amyloid, heart
The vessel walls everywhere are often heavily involved, though infarcts are unusual.
Both amyloid A and amyloid B involve vessels heavily (Mod. Path. 3: 4, 1990).
{3386} amyloid, coronary artery (left: Sirius stain;
right: crystal violet)
Amyloid-laden vessels are brittle. This produces problems ranging from trivial "post-proctoscopy
palpebral purpura" to dread intracranial hemorrhage (fortunately rare in the common systemic
amyloidosis syndromes).
GI tract (Dig. Dis. Sci. 15: 155, 1997)
Stiff tongue is a well-known problem in both systemic (B) and localized (organ-specific)
amyloidosis.
Amyloid deposits around the blood vessels of the gut result in malabsorption.
Amyloid in the ganglia and wall of the gut makes normal peristalsis impossible.
{24551} amyloid in small bowel (vessels and outer layer of muscularis propria)
Liver
Amyloid is deposited first in the perisinusoidal spaces, and this appears to crunch the hepatocytes
after a while.
However, because the sinusoids remain open and unscrambled,
the liver seldom fails, and pressure in the portal vein usually remains normal.
Although it has long been taught that liver biopsy is very dangerous
if amyloidosis is suspected (i.e., the amyloid holds the sinusoids wide-open,
inviting massive hemorrhage), Mayo's recently found the danger isn't real.
Series Medicine 82: 291, 2003; also confirms the poor prognosis.
{39805} amyloid in the liver
Adrenal gland
Amyloidosis can and does present as adrenal cortical insufficiency ("Addisonism").
{24607} amyloid, adrenal (looks and feels like candle wax)
Bleeding tendency
First problem: amyloid deposited around small blood vessels renders them more fragile.
Thus, problems similar to those seen in scurvy result. Look for little hemorrhages in the tissues
around the eyeballs.
*Second problem: amyloid B can soak up coagulation factor X,
requiring plasma exchange
(Am. J. Haem. 54: 68, 1997).
Spleen
Amyloid deposition in the spleen is usual in systemic amyloidosis.
Most patients get increased circulating platelets, but the relative loss of splenic function is among the
least of these peoples' problems.
A "sago spleen" has its amyloid in the white pulp. (Sago is tapioca.) A "lardaceous spleen" has its
amyloid in the red pulp (* and usually indicates amyloid B -- see Mod. Path. 3: 419, 1990). It looks
like lard. (The distinction is of no practical importance, but every physician seems to remember it!)
Skin
The skin may be involved trivially in generalized disease.
{12215} amyloid, eyelids
*Several minor skin diseases feature local accumulations of an amyloid
derived from a keratin
("amyloid D").
Nerves
Some weakness and loss of sensation is common in amyloidosis, with or without unpleasant
sensations. (* This is especially common in hereditary amyloidosis syndromes. It also occurs early
in amyloidosis B -- be suspicious.) Morphology Arch. Path. Lab. Med. 124:
114, 2000.
Wrist
Carpal tunnel syndrome (too-tight flexor retinaculum) developing in an older person makes the
astute clinician suspect amyloidosis. (* Check Tinel's sign.)
Lungs
At least three patterns occur: nodules (usually asymptomatic),
diffuse interstitial infiltration (dyspnea), and tracheobronchial amyloidosis
(obstructing the larger airways; Medicine 79: 69, 2000; treatment with the new external-beam machine
Mayo Clin. Proc. 76: 853, 2001).
Making the diagnosis
To diagnose amyloidosis, you must send the lab some solid tissue and the pathologist must find
amyloid in it.
*(Don't try the old intravenous Congo Red test on your patients. Allergic responses are common.)
If the amyloid is discovered on biopsy of kidney, liver, or other organ, you have the
diagnosis. (This is often a big surprise.)
Your pathologist can distinguish the common amyloids histochemically.
Be suspicious that your older patients have amyloidosis. ("Amyloidosis is in the differential
diagnosis of just about every symptom".)
Current practice is to cut off a piece of gingival or rectal tissue and send it to the pathologist. It is
polite and helpful to mention you are looking for amyloid.
Such a biopsy will demonstrate amyloid ("the biopsy is positive") in majority of advanced cases.
Negative biopsy does not exclude systemic amyloidosis!
Aspiration of subcutaneous fat is an okay way to start looking for amyloid (Am. J. Med. 82: 412,
1987 -- detects the common amyloids but not amyloid H). * Gastric biopsy is another high-yield
procedure (Hum. Path. 16: 1206, 1986).
If you find amyloidosis, you may want to check the patient over for some underlying disorder (i.e.,
B-cell neoplasm, chronic infection). If you find something treatable, you can slow down, but not
reverse, the amyloid deposition.
Treatment and prognosis
We wish we had more to offer people
with systemic amyloidosis.
Untreated patients with amyloidosis B usually die around one year after diagnosis from heart problems,
but about 5% make it to ten years with today's chemotherapy (Blood 93: 1062, 1999).
Patients with amyloidosis A usually die around ten years after diagnosis from kidney failure.
Some specific ways of helping do exist.
Aggressive treatment of amyloidosis B (Hosp. Pract. 31(8):
67, Aug 15, 1996)
is becoming
popular. Options include bone marrow transplantation (Br. J. Haem. 100: 229,
1998 & 101: 766, 1998),
super-intense melphalan plus stem cell support (Blood 91: 3662, 1998; Am. J. Med. 113:
549, 2002),
and alfa-interferon.
The treatment of amyloidosis A is that of the underlying disease. Occasionally the amyloid
regresses when the underlying disease is cured. But don't count on it.
Cardiac amyloidosis causing heart block can be treated by inserting an electrical pacemaker to keep
the ventricles beating properly.
Most hereditary amyloidosis syndromes require genetic counselling.
Congophilic angiopathy will result in intracranial bleeding regardless of any treatment.
*Watch for undiscovered, relative non-toxic anthracyclines that will bind to the beta-pleated sheets,
render them more soluble and prevent additional amyloid from being laid down on them. Such
anthracyclines are available now, but unfortunately they're deadly poison.
INTRODUCTION TO IMMUNODEFICIENCY
The immunodeficiency states are hereditary, infectious, and iatrogenic disorders. Patients have extra
trouble fighting infections.
Systemic diseases that result in immunodeficiency include alcoholism, diabetes, nephrotic syndrome,
uremia, and Cushing's syndrome. Cancer is associated by immune suppression by a variety of
mechanisms. Malnutrition is another important cause of immunodeficiency. Immunosuppressive
drugs are often given to treat autoimmune disease or cancer, or to halt transplant rejection. More
about all this later.
In immunodeficiency states that might be due to some mysterious T-cell defect (including all the
mysterious B-cell defects), patients generally have a high chance of developing "autoimmune
diseases" of various sorts.
Patients with primary deficits in cell-mediated immunity almost all have oral candidiasis
(thrush,
yeast infection), and go on to have problems with intracellular parasites
like CMV and pneumocystis. Bacterial infections are likely to be
sepsis rather than localized. If severe, these will show up soon after birth.
Patients with primary deficits in humoral
immunity
have most problems with pyogenic bacteria (staph, pneumococcus, H. 'flu,
etc.) These won't show for the first few months of life because of Mom's antibodies.
Neutrophil problems
include such illnesses as chronic granulomatous disease, Chediak-Higashi
disease, adhesion molecule problems,
etc., etc. that are not part of today's unit. Expect problems
especially with staph, pseudomonas, candida, nocardia, and aspergillus.
Watch for more immune problems as the bioengineered "biological response
modifiers* gain popularity. The TNF-binders infliximab and etanercept,
while obviously powerful and useful, have caused opportunistic infections,
especially by mycobacteria (NEJM 345: 1098, 2001) and fungi.
When tumor necrosis
Patients with defects in some of the complement components
involved in membrane attack (C5, C6, C7, C8, also properdin; C9 is asymptomatic)
are especially troubled with neisseria.
Patient with defects in C2 or C4 are likely to have a lupus-like syndrome
(butterfly rash, arthritis, nephritis). The reason why remains mysterious,
and the disease is bad (Arch. Derm. 136: 1508, 2000).
Patients with spleen loss (sicklers, post-surgery)
have particular problems with salmonella, pneumococci, * listeria,
and * DF2 dogbite bacterium.
HEREDITARY IMMUNODEFICIENCIES ("primary immunodeficiencies"; reviews
Ped. Clin. N.A. 47(6): Dec. 2000, whole issue; Mayo Clin. Proc. 73: 865, 1998;
J. Allerg. Clin. Imm. 111(2S): S-571, 2003.
molecular diagnosis of 75 of the ~100 known syndromes is now
available, review Lancet 357: 1863, 2001; Nat. Imm. 5: 23, 2004)
X-linked agammaglobulinemia (Bruton's disease, etc.)
The B-cells are absent, fail to mature, or at least fail to respond to infection. The lymph nodes are
tiny, without germinal centers.
Molecular biology: Science 261: 355 & 358, 1993;
J. Imm. 161: 3925, 1998. The deficiency
is in Bruton's B-cell progenitor tyrosine kinase (btk,
Cell 72: 279, 1993), which is a gene that tells B-cells to multiply
when they are stimulated.
More on the gene:
Nature 361: 226, 1993; alleles Pediatrics 101:
276, 1998.
All classes of immunoglobulins are absent or nearly absent. When mother's immunoglobulins are
gone (six months after birth), severe pyogenic bacterial infections begin.
The disease is controlled more or less successfully
with injections of gamma globulin.
*Survivors have high prevalence of lupus, polymyositis-dermatomyositis, polyarteritis, rheumatoid
arthritis, etc.
*Don't give these kids live polio vaccine.
Primary care physicians: These patients present with early and intractable
otitis media. You'll be criticized if you don't check
these people (J. Ped. 41: 566, 2002).
Isolated IgA deficiency ("selective IgA deficiency")
"The commonest immunodeficiency syndrome", affecting around one in maybe 300
people (* estimates vary.)
No one understands the cause; there's a familial tendency
but no obvious pattern of inheritance (update J. Immuno. 169:
4637, 2002).
Serum and secretory IgA are very low or absent. * There is disagreement about whether these
patients have "more colds" and "more GI infections", or "have worse allergies", or "are more likely
to get collagen-vascular disease", etc., etc.
The most serious hazard is iatrogenic anaphylaxis from the second administration of blood plasma.
*Isolated IgM deficiency also occurs but is much less common than isolated IgA deficiency. Yet
other people lack certain subclasses of IgG.
* Transient hypogammaglobulinemia of infancy features some extra
infections, with a delay in producing some antibody subclass (usually
IgG2). These children
do produce antibodies in response to challenge immunization, and usually
no treatment is required.
DiGeorge's Syndrome ("thymic dysembryogenesis")
The third and fourth branchial pouches fail to form properly. While the patient is being treated for
tetany and cardiac malformations just after birth, the lack of T-cells becomes evident as a fungal,
viral, or other infection.
* It is not familial.
Most of these kids have monosomy 22q11 (Am. J. Hum. Genet. 51: 964, 1992).
A graft of fetal thymus can be very helpful to some of these patients --
even restorative (NEJM 341: 1227, 1999). But the best seems to be a HLA-matched
thymus transplant (Blood 102: 1121, 2003). Update Blood 104:
2574, 2004.
*"Nezelof's syndrome" is a possibly related syndrome; these patients have very little thymic tissue
but normal parathyroid hormone levels; some of these patients lack purine nucleoside
phosphorylase.
Severe combined immunodeficiency (SCID) syndromes
A variety of profound deficiencies of both T-cell and B-cell function.
Genetics update J. Clin. Inv. 114: 1409, 2004.
There are
several autosomal
SCID syndromes.
Perhaps half of autosomal-recessive SCID are due to adenosine deaminase deficiency or another
(J. Ped. 128: 373, 1996)
defect in purine salvage. The problem is that too much adenosine builds up, and gets turned into
dATP, which is very poisonous for all lymphocytes. Or maybe the problem is that adenosine
deaminase, which normally binds to CD26, the T-cell activation
receptor, makes it work; Science
261: 466, 1993, adult alleles Blood 89: 2849, 1997.
HLA-matched bone marrow transplant is curative or....
ADA deficiency was the first disease to be cured by introduction of the normal
gene into cells. Beginnings: MWN 9/22/86, p. 42; outcome Br. Med. J. 304: 1202, 1992; Science
258: 744, 1992 (both girls are now healthy and in public schools).
In "bare lymphocyte disease", macrophages fail to
express HLA-II antigens.
* CD45 deficiency: Nat. Med. 6: 343, 2000.
* Deficiencies of the RAG-1 or RAG-2 genes ("recombination activating")
that produce lymphocyte diversity cause a severe combined
immunodeficiency; there is a forme fruste (Blood 06: 2099, 2005).
At least three X-linked combined immunodeficiency are known.
In one form of X-linked SCID (*SCIDX1), the interleukin 2
receptor (* gamma chain, IL2RG, which it shares with the
interleukin 4 and 7 receptors) is defective (Blood 83: 626, 1994; Science 263: 1453, 1994; Science
262: 1887 & 1880, 1993; Blood 88:
1708, 1996, J. Clin. Invest. 99:
160, 1997).
This affected David
Veter "the bubble boy". See JAMA 251: 1929 and 1935,
1984; JAMA 253: 74 & 78, 1985 for this shameful fiasco.
In one recent
series, all 12 SCID children whose biochemistry remained obscure were boys,
telling me that all the major autosomal SCID loci have been found: J. Ped. 130:
378, 1997. * New one with mutated CD45: Nat. Med. 343: 2000.
Gene therapy succeeds for SCID-X1: Science 288: 669, 2000; NEJM 346: 1185, 2002.
*Another autosomal recessive form has been found to result from deficient
Jak3 protein kinase,
which binds to the IL2-R gamma chain cited above (Nature 37: 65, 1995; Science 270: 797, 1995;
Blood 90: 3996, 1997;
Blood 91: 949, 1998).
Yet another is the human equivalent of the nude mouse (Blood 97: 800, 2001).
And there are at least two interferon receptor deficiencies now known
(Nat. Genet. 33: 388, 2003).
*The white cells in a single blood transfusion will give SCID patients graft-versus-host
disease, and this can even happen from the maternal lymphocytes
that enter the child's bloodstream during childbirth.
Ataxia-telangiectasia (* "Louis-Bar syndrome",
NEJM 325: 1831, 1991; Medicine 70: 99, 1991; QJM 82(299): 197, 1992)
A poorly-understood, autosomal-recessive (* probably several alleles on chromosome 11) systemic
problem involving brain (* lack of Purkinje cells), vessels, and the immune system. Something is
wrong with gene expression and tissue differentiation. The disease is inherited as an autosomal
recessive *on 11q22-23 -- gene cloned Science 268: 1749, 1995; NEJM 333: 777, 1995), and
patients have fragile chromosomes, with notable break points at sites involved in T-cell receptors
and immunoglobulins.
*Interestingly, most of the recombination events take place within, rather than between, individual
chromosomes (Science 260: 1327, 1993.)
AT heterozygotes (around 0.5% of humankind) have poor tolerance for radiation therapy; they may
need to be distinguished from other patients in designing protocols, establishing doses, etc. They
also have a clear increase (3.5 x others) in cancer risk. Stay tuned for improved identification of
these unfortunates.
Cellular immunity against viruses is poor, and many of these patients cannot make IgA or IgE.
Patients eventually die from lung infections or cancer.
{53740} ataxia-telangiectasia, patient (all you can tell from this picture is this is some kind of
nervous system disease)
Wiskott-Aldrich syndrome ("immunodeficiency with thrombocytopenia and eczema";
Blood 103: 456, 2004)
Deficiency (usually) of WASP, an important but poorly-understood protein.
It is as an X-linked recessive trait.
Update on the alleles: J. Immuno. 175: 1329, 2005.
Affected boys have eczema, low platelet counts and volumes
(they are fragile and break in the ciculation: Blood 94: 509, 1999),
and repeated infections associated with
variable losses of cellular immunity (especially against viruses) and/or hypercatabolism of
immunoglobulins (mostly affecting IgM).
The prognosis is poor unless treatment with bone marrow transplantation is successful (Blood 82:
2961, 1993, lots more). If no donor is available, splenectomy (to increase platelet counts) can add several
good years, and cord blood from unrelated donors offers hope as well (J. Ped. 142: 519, 2003).
There is a moderate increase in lymphomas, perhaps due to hyperplasia of the lymphocytes.
* Chromosomes are stable in this disorder.
* Another allele gives only thrombocytopenia: Blood 90:
2680, 1997.
* About all we know so far about WASP is that it enables expression
of CD43 on all lymphocytes.
Sex-linked lymphoproliferative syndrome
Boys who are unable to deal with the Epstein-Barr virus ("infectious mononucleosis virus"), and
develop lymphoma as a result. (*Future pathologists: Erythrophagocytosis.)
The gene is SH2D1A and the protein is under study:
Nat. Genet. 20; 129, 1998, PNAS 95: 13765, 1998.
* T-cell membrane defects (J. Allerg. Clin. Imm. 109: 747, 2002)
A group of hereditary syndromes that have only recently been characterized. They are described
well in Big Robbins.
Mutant CD3-gamma subunit, the first defective T-cell marker syndrome: NEJM 327: 529, 1992.
* Thankfully rare: Idiopathic CD4+ T-lymphopenia ("HIV-negative
AIDS" is a term that shouldn't be used.) The CD4+ T-cells
undergo apoptosis; nobody knows why (J. Clin. Invest. 97:
672, 1996).
* Thankfully rare: Bare lymphocyte disease
is lack of MCH II proteins: NEJM 337: 748, 1997;
J. Immunol. 158: 5841, 1997,
J. Immunol. 159: 1086, 1997 (gene rx trial);
lesions at any of several different known loci can cause it Blood 100:
1496, 2002.
COMMON VARIABLE IMMUNODEFICIENCY ("acquired hypogammaglobulinemia" --
update Mayo Clin. Proc. An unclassifiable group of syndromes that can begin in anyone at any age, but mostly in young
adults.
As you'd expect,
the patients suffer from recurrent bacterial infections and * giardiasis.
B-cells are present in normal numbers, but fail to turn into plasma cells as they should. * Gluten
enteropathy and autoimmune diseases are common.
Often, there is tremendous hyperplasia of the useless germinal centers, simulating "nodular
malignant lymphoma".
In one variant (formerly in the "CVI" wastebasket), the patients make lots of IgM but little of any other immunoglobulin (NEJM 319:
495, 1988). When this is X-linked (X-linked hyper IgM syndrome),
the cause is a defective
gene for CD40 ligand (Science 259: 990,
1993; NEJM 330:
949, 1994, alleles J. Ped. 131: 47, 1997.).
Still other patients simply have bad alleles of C2, or the T-helper cells don't produce much IL-2 or
gamma-interferon, or a forme-fruste of x-linked lymphoproliferative
disease (Blood 98: 1321, 2001) or whatever.
You can guess what lesions might be found.
Update on the T-cell-based CVI's: Blood 106: 626, 2005.
* Another has mutant α-TNF and lots of granulomas
(J. Imm. 159: 6236, 1997).
Yet another group (hyper-IgE syndrome, also recently removed from the CVI wastebasket)
has lots of IgE, severe deficiency in the other immunoglobulins, and some T-cell
problems too. This is called "Job's syndrome", after the hero of the Old Testament book who
suffered a serious of disasters culminating in necrotizing skin infections.
* Regardless of your beliefs
or background, you'd enjoy reading the whole "Book of Job" through sometime, especially if you
dislike the facile wrong answers of certain "ministries"; it's not what you've been told.
*Five % of patients with primary tumors of the thymus
have their cells stop making most antibodies
("Good's hypogammaglobulinemia"; South. Med. J. 90:
444, 1997, still a mystery).
GENETIC DEFICIENCIES OF THE COMPLEMENT SYSTEM
Hereditary angioedema (C1-esterase inhibitor deficiency, not really an "immunodeficiency")
An autosomal dominant deficiency of an inhibitor of the early phase of classical complement
activation. (Molecular biology of the gene: NEJM 317: 1 & 43 & 641, 1987). Now that most
physicians are alert to this entity, more and more cases are being discovered.
The patient suffers dramatic edema (but not urticaria) in various parts of the body, unpredictably,
following minor insults. Angioedema of the larynx occasionally causes sudden death in these
patients.
*Acquired C1-esterase inhibitor deficiency develops (rarely) in malignant lymphoma patients and in
the presence of autoantibodies against the factor (NEJM 316: 1360, 1987).
The era of undertreating this entity is probably over. A concentrate of the inhibitor is now available:
NEJM 334: 1630, 1996.
Complement component deficiency syndromes: increased risk for infection
(notably neisserial
infections) and/or systemic lupus-like illness.
C2 deficiency, the most common, presents an anti-nDNA-negative lupus-like picture.
*C4 deficiency or variants (the two loci are highly pleomorphic), also gives a lupus-like picture (J.
Rheum. 18: 345, 1991).
* Mannose-binding lectin deficiency (MBL deficiency): A newly discovered genetic defect that
renders people more susceptible to various infections, including HIV (Lancet 349: 236, 1997).
* The pop claim that "too many vaccines overwhelm and weaken the immune system"
examined: Pediatrics 109: 124, 2002. As with most other things, non-scientifically-minded
people will base their beliefs on emotion. Unlike most other beliefs, falling for this
stuff can have serious consequences for the children and the community.
If people are aware that you've examined this claim and have dismissed it
(and especially if they realize that the anti-immunization activists are bunko artists),
most of them will accept your support of immunization.
* Amyloidosis
You are about to become very rubbery.
Flesh setting like a bowl of cooling jello,
How it dyes my slide
O pernicious microsplinters!
Ed says, "This world would be a sorry place if
people like me who call ourselves Christians
didn't try to act as good as
other
good people
."
Prayer Request
COMPOSITION OF AMYLOID
AMYLOID NAME
CLINICAL CORRELATIONS
Serum amyloid associated protein (acute phase reactant)
Amyloid A (AA)
chronic inflammation (leprosy, osteomyelitis, tuberculosis,
rheumatoid arthritis, etc.); New Guinea amyloid goiter
Immunoglobulin light chains
Amyloid B (AL)
B-cell proliferations (plasma cell myeloma, benign hyperplasias)
Transthyretin ("prealbumin")
Amyloid C (AF)
Substituted: Familial syndromes (Amyloid ATTR)
Various endocrine proteins
Amyloid Esub
Stromas of endocrine tumors (various ones)
Beta-two microglobulin
Amyloid H
Chronic hemodialysis patients
Amyloid beta protein
Amyloid beta / A4
Cores of Alzheimer senile plaques; cerebral blood
vessels in Alzheimer's and some congophilic
angiopathy patients
Tau microtubule protein
tangles, plaques, and granules of Alzheimer's
Amylin (islet amyloid-associated peptide)
stroma of insulinomas; islands of some type II diabetics
Prolactin
(An amyloid E)
Old folks' pituitaries
Atrial natriuretic peptide/factor
A-ANF
Old folks' atria
Keratin
Amyloid D
Some skin diseases
Gamma trace alkaline microprotein (cystatin C)
Hereditary congophilic angiopathy of Iceland
Gelsolin
AGel
Familial Finnish (Am. J. Path. 126: 1223, 1990)
Prion Precursor Protein (PRPP)
AScr
Prion diseases
Apolipoprotein A1 (Am. J. Path. 154: 221, 1999)
AApoA1
Familial Iowa amyloid; dog amyloid
Amyloid
Congo red stain
WebPath Photo
Amyloid
Bryan Lee
Amyloidosis
Nice discussion
Rockford Case of the Month
Amyloidosis
Lots of apple green birefringence
Wash U, St. Louis
{08132} Congo red, birefringence
{35978} crystal violet stain, rectal positive for amyloid (red)
Amyloid
Electron micrograph
WebPath photo
{08165} amyloid, histology, glomeruli
{07937} amyloid between myocardial cells, histology
{11021} amyloid in the kidney, thioflavin T (stains amyloid yellow and everything else black)
{16664} amyloid, electron micrograph
{17334} amyloid, electron micrograph
Renal Amyloidosis
Electron micrographs
VCU Pathology
* Savvy cardiologists now biopsy the subcutaneous fat at time of
pacemaker implantation, and send it to the pathologist to be checked
for amyloid: Heart 87: 7, 2002.
{22023} amyloid, cornea
{22026} amyloid, cornea, histology
{22029} amyloid, conjunctival surface, histology
Amyloidoma
Lung pathology series
Dr. Warnock's Collection
{10883} renal amyloidosis, congo red
{11021} renal amyloidosis, thioflavin T
{16785} amyloid in the glomerulus
{17171} amyloid in the glomerulus
 Amyloidosis
Amyloidosis
WebPath Photo
{24519} amyloid heart, gross (looks and feels like candle wax)
{10685} congo red stain (my case)
{10688} congo red, polarized (my case again)
{10370} amyloid in gut wall (Congo red and unstained slices)
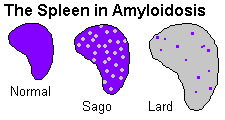 There are two classic, unappetizing descriptions of amyloid-loaded spleens seen at autopsy.
There are two classic, unappetizing descriptions of amyloid-loaded spleens seen at autopsy.
Sago spleen
Urbana Atlas of Pathology
4'-iodo-4'-deoxy-doxorubicin (IDX) for prions: Science 276: 1119, 1997.
More: Drugs & Aging 8: 75, 1996.
Less often, the mutation is on the mu heavy chain gene
(NEJM 335: 1486, 1996).
DiGeorge syndrome
Pittsburgh Pathology Cases
As a result, there are no T-cell and no NK-cells.
In 2002, one boy in France developed leukemia with the retroviral vector found to
be inserted next to an oncogene. The FDA banned the technique even though
it was an obvious life-saver for other kids,
while the British talked (more sensibly, I think) about benefits vs. risks
(Nature 419: 545, 2002; Br. Med. J. 325: 791, 2002).
The disease in inherited as an autosomal dominant
with variable expressivity. The baby
teeth don't shed on time (or sometimes at all). NEJM 340:
692, 1999).
(to the Patient)
I see it in my microscope:
Mafioso smothered in polymer.
A rainbow that possesses the iris
And quells a life,
Engulfs and annihilates,
Transforms organs into waxy casts
Into incandescent resins.
Celestial spectrum,
And your insides --
Brimming with sweet peach,
Fire of violet or apple-green.
An organic pallet of dry sun showers,
It is gaudy as the devil or a gas chromatograph.
Pricks the innocent interstitium.
Soon you will bounce like a ball,
Flash like a hall full of candles.
-- Sheila M. Katz, M.D.
Professor of Pathology
Hahnemann University, Philadelphia
Medical Heritage 1: 299, 1985.

Visitors to www.pathguy.com
reset Jan. 30, 2005: Teaching Pathology
Teaching Pathology
PathMax -- Shawn E. Cowper MD's
pathology education links
Ed's Autopsy Page
Notes for Good Lecturers
Small Group Teaching
Socratic
Teaching
Preventing "F"'s
Classroom Control
"I Hate Histology!"
Ed's Physiology Challenge
Pathology Identification
Keys ("Kansas City Field Guide to Pathology")
Ed's Basic Science
Trivia Quiz -- have a chuckle!
Rudolf
Virchow on Pathology Education -- humor
Curriculum Position Paper -- humor
The Pathology Blues
 Ed's Pathology Review for USMLE I
Ed's Pathology Review for USMLE I
![]()
![]()
Pathological Chess

Taser Video
83.4 MB
7:26 min

 Polyarteritis Nodosa
Polyarteritis Nodosa
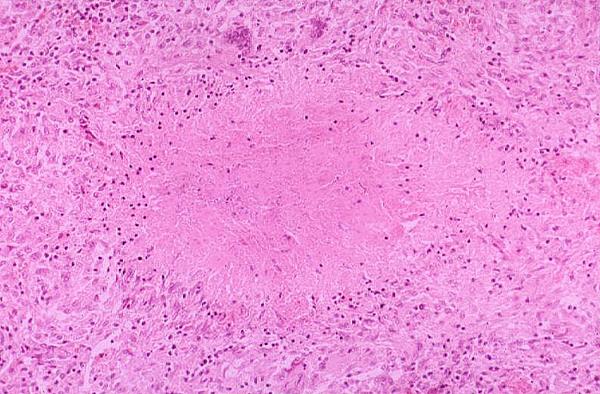 Granuloma
Granuloma)window.location='http://www.mdchoice.com/photo/img/img0049.jpg') Angioedema
Angioedema
