Ed Friedlander, M.D., Pathologist
scalpel_blade@yahoo.com



Cyberfriends: The help you're looking for is probably here.
Welcome to Ed's Pathology Notes, placed here originally for the convenience of medical students at my school. You need to check the accuracy of any information, from any source, against other credible sources. I cannot diagnose or treat over the web, I cannot comment on the health care you have already received, and these notes cannot substitute for your own doctor's care. I am good at helping people find resources and answers. If you need me, send me an E-mail at scalpel_blade@yahoo.com Your confidentiality is completely respected.
 DoctorGeorge.com is a larger, full-time service.
There is also a fee site at myphysicians.com,
and another at www.afraidtoask.com.
DoctorGeorge.com is a larger, full-time service.
There is also a fee site at myphysicians.com,
and another at www.afraidtoask.com.
Translate this page automatically
 |
With one of four large boxes of "Pathguy" replies. |
 I'm still doing my best to answer
everybody.
Sometimes I get backlogged,
sometimes my E-mail crashes, and sometimes my
literature search software crashes. If you've not heard
from me in a week, post me again. I send my most
challenging questions to the medical student pathology
interest group, minus the name, but with your E-mail
where you can receive a reply.
I'm still doing my best to answer
everybody.
Sometimes I get backlogged,
sometimes my E-mail crashes, and sometimes my
literature search software crashes. If you've not heard
from me in a week, post me again. I send my most
challenging questions to the medical student pathology
interest group, minus the name, but with your E-mail
where you can receive a reply.
Numbers in {curly braces} are from the magnificent Slice of Life videodisk. No medical student should be without access to this wonderful resource. Someday you may be able to access these pictures directly from this page.
Also:
Medmark Pathology -- massive listing of pathology sites
Freely have you received, freely give. -- Matthew 10:8. My
site receives an enormous amount of traffic, and I'm
handling about 200 requests for information weekly, all
as a public service.
Pathology's modern founder,
Rudolf
Virchow M.D., left a legacy
of realism and social conscience for the discipline. I am
a mainstream Christian, a man of science, and a proponent of
common sense and common kindness. I am an outspoken enemy
of all the make-believe and bunk that interfere with
peoples' health, reasonable freedom, and happiness. I
talk and write straight, and without apology.
Throughout these notes, I am speaking only
for myself, and not for any employer, organization,
or associate.
Special thanks to my friend and colleague,
Charles Wheeler M.D.,
pathologist and former Kansas City mayor. Thanks also
to the real Patch
Adams M.D., who wrote me encouragement when we were both
beginning our unusual medical careers.
If you're a private individual who's
enjoyed this site, and want to say, "Thank you, Ed!", then
what I'd like best is a contribution to the Episcopalian home for
abandoned, neglected, and abused kids in Nevada:
My home page
Especially if you're looking for
information on a disease with a name
that you know, here are a couple of
great places for you to go right now
and use Medline, which will
allow you to find every relevant
current scientific publication.
You owe it to yourself to learn to
use this invaluable internet resource.
Not only will you find some information
immediately, but you'll have references
to journal articles that you can obtain
by interlibrary loan, plus the names of
the world's foremost experts and their
institutions.
Alternative (complementary) medicine has made real progress since my
generally-unfavorable 1983 review linked below. If you are
interested in complementary medicine, then I would urge you
to visit my new
Alternative Medicine page.
If you are looking for something on complementary
medicine, please go first to
the American
Association of Naturopathic Physicians.
And for your enjoyment... here are some of my old pathology
exams
for medical school undergraduates.
I cannot examine every claim that my correspondents
share with me. Sometimes the independent thinkers
prove to be correct, and paradigms shift as a result.
You also know that extraordinary claims require
extraordinary evidence. When a discovery proves to
square with the observable world, scientists make
reputations by confirming it, and corporations
are soon making profits from it. When a
decades-old claim by a "persecuted genius"
finds no acceptance from mainstream science,
it probably failed some basic experimental tests designed
to eliminate self-deception. If you ask me about
something like this, I will simply invite you to
do some tests yourself, perhaps as a high-school
science project. Who knows? Perhaps
it'll be you who makes the next great discovery!
Our world is full of people who have found peace, fulfillment, and friendship
by suspending their own reasoning and
simply accepting a single authority that seems wise and good.
I've learned that they leave the movements when, and only when, they
discover they have been maliciously deceived.
In the meantime, nothing that I can say or do will
convince such people that I am a decent human being. I no longer
answer my crank mail.
This site is my hobby, and I presently have no sponsor.
This page was last updated February 6, 2006.
During the ten years my site has been online, it's proved to be
one of the most popular of all internet sites for undergraduate
physician and allied-health education. It is so well-known
that I'm not worried about borrowers.
I never refuse requests from colleagues for permission to
adapt or duplicate it for their own courses... and many do.
So, fellow-teachers,
help yourselves. Don't sell it for a profit, don't use it for a bad purpose,
and at some time in your course, mention me as author and KCUMB as my institution. Drop me a note about
your successes. And special
thanks to everyone who's helped and encouraged me, and especially the
people at KCUMB
for making it possible, and my teaching assistants over the years.
Whatever you're looking for on the web, I hope you find it,
here or elsewhere. Health and friendship!
 I am presently adding clickable links to
images in these notes. Let me know about good online
sources in addition to these:
I am presently adding clickable links to
images in these notes. Let me know about good online
sources in addition to these:
Pathology Education Instructional Resource -- U. of Alabama; includes a digital library
Houston Pathology -- loads of great pictures for student doctors
Pathopic -- Swiss site; great resource for the truly hard-core
Syracuse -- pathology cases
Walter Reed -- surgical cases
Alabama's Interactive Pathology Lab
"Companion to Big Robbins" -- very little here yet
Alberta
Pathology Images --hard-core!
Cornell
Image Collection -- great site
Bristol Biomedical
Image Archive
EMBBS Clinical
Photo Library
Chilean Image Bank -- General Pathology -- en Español
Chilean Image Bank -- Systemic Pathology -- en Español
Connecticut
Virtual Pathology Museum
Australian
Interactive Pathology Museum
Semmelweis U.,
Budapest -- enormous pathology photo collection
Iowa Skin
Pathology
Loyola
Dermatology
History of Medicine -- National Library of Medicine
KU
Pathology Home
Page -- friends of mine
The Medical Algorithms Project -- not so much pathology, but worth a visit
National Museum of Health & Medicine -- Armed Forces Institute of Pathology
Telmeds -- brilliant site by the medical students of Panama (Spanish language)
U of
Iowa Dermatology Images
U Wash
Cytogenetics Image Gallery
Urbana
Atlas of Pathology -- great site
Visible
Human Project at NLM
WebPath:
Internet Pathology
Laboratory -- great site My team:
My team:Ed Lulo's Pathology Gallery
Bryan Lee's Pathology Museum
Dino Laporte: Pathology Museum
Tom Demark: Pathology Museum
Dan Hammoudi's Site
Claude Roofian's Site
Pathology Handout -- Korean student-generated site; I am pleased to permit their use of my cartoons
Estimating the Time of Death -- computer program right on a webpage
Pathology Field Guide -- recognizing anatomic lesions, no pictures

St.
Jude's Ranch for Children
I've spent time there and they are good. Write "Thanks
Ed" on your check.
PO Box 60100
Boulder City, NV 89006--0100
More of my notes
My medical students
Clinical
Queries -- PubMed from the National Institutes of Health.
Take your questions here first.
HealthWorld
Yahoo! Medline lists other sites that may work well for you

We comply with the
HONcode standard for health trust worthy
information:
verify
here. 

![]()
Distinguish acute pancreatitis and "chronic pancreatitis", and recognize the terms that describe the severity of the former illness.
Describe the typical clinical settings for acute pancreatitis, its presentation, and what is known of its etiology.
Distinguish the two histologic types of chronic pancreatitis, and their differing etiologies. Describe and recognize the histopathology of both.
Cite the known and possible risk factors for cancer of the pancreas. Describe its typical presentation, anatomic pathology, and course. Name the mutation most strongly linked to this disease, and the operation which is occasionally curative.
Define "diabetes mellitus", "impaired glucose tolerance", "gestational diabetes mellitus", and "previous/potential abnormality of glucose tolerance". Mention some archaic synonyms for each one. Tell when hyperglycemia simply isn't diabetic.
Describe the essential lesion in, and typical clinical course of, type I diabetes. Outline current thinking about its etiology, cite the risk to siblings and twins, and describe the HLA association.
Describe the essential lesion in, and typical course of, type II diabetes. Describe current thinking about the pathophysiology of this illness. Mention its genetics. Identify MODY, its most familiar genetic locus, and its pattern of inheritance.
Explain the pathophysiology of diabetic ketoacidosis and hyperosmolar nonketotic coma.
Define "secondary diabetes mellitus". Recognize the important causes. Compare the effects of hyperglycemia on the rest of the body in secondary diabetes and primary diabetes. Briefly describe amylin.
Tell why diabetics have increased polyols, and relate this to complications.
Distinguish diabetic large and small vessel disease. Suggest why diabetics so often lose legs. Outline the common renal lesions in diabetes.
Identify the causes of blindness in diabetes. Give the anatomic pathology of the various forms of diabetic retinal disease.
Describe the things that happen to the peripheral nerves of diabetics, and what problems these cause.
Explain what is meant by "nonenzymatic glycosylation". Tell how this relates to thinking about diabetic complications, and to the HgbA1c blood test for diabetic control.
Describe insulin shock, fasting hypoglycemia, and postprandial hypoglycemia. Give a simple differential diagnosis for the last two. Tell what really causes the "idiopathic postprandial syndrome".
Comment on the following, heard at a party: "Diabetes is caused by eating refined sugar. If there were no white sugar, there would be no diabetes. There should be a law!"
Recognize the following histopathologic lesions of diabetes: diabetic nodular glomerulosclerosis, diabetic arteriolar sclerosis, hepatic nuclear glycogenosis, and hyalinization (amyloid/collagen) of islets.
QUIZBANK Disk 3: Metabolic #'s 42-81 Disk 7: Pancreas (all except #'s 1-8)
|
|
|
|
|
NORMAL PANCREAS
I'm tired of all this nonsense about beauty being only skin-deep. That's deep enough. What do you
want -- an adorable pancreas?
Eat when you can.
Sleep when you can.
Don't touch the pancreas.
 {25019} normal pancreas, the white hamburger
{25019} normal pancreas, the white hamburger
{14887} normal pancreas, trichrome; no blue, so no
dense fibrous tissue in the healthy pancreas.
{12463} islets of Langerhans (no, IZZ-lett is not really an acceptable pronunciation);
H&E stain
Unqualified, disease of the pancreas ("pancreatitis", "pancreatic tumor", "pancreatic cancer", etc.) means disease of the exocrine pancreas. The plural of "pancreas" is "pancreata".
 You are already familiar with the gross and microscopic structure, and the physiology, of the exocrine
and endocrine pancreas.
You are already familiar with the gross and microscopic structure, and the physiology, of the exocrine
and endocrine pancreas.
Because of its lobulated texture, many surgeons and pathologists call the pancreas "the hamburger".
Remember that the islands make up about 15% of the organ's weight, and that they are concentrated in the tail.
Fatty ingrowth is common in the normal pancreas, and this fat can undergo fat necrosis.
The exocrine pancreas has great functional reserve, and no symptoms will occur until 85% or so of the gland is gone.
To keep your pancreas healthy, your digestive enzymes must not be activated until they reach the duodenum! Ordinarily, trypsinogen is activated only on contact with enterokinase (from the duodenal mucosa), and trypsin in turns activates the other enzymes.
* Future pathologists: Immunostaining the cells of the pancreas (Cancer 66: 2134, 1990):
acinar... trypsin, lipase
ductal... carbonic anhydrase, others
islet... somatostatin, chromogranin
The embryonic pancreas arises from the duodenum as two buds.
* Embryologists: The "dorsal bud" contributing the bulk of the organ, all that is drained by the duct of Wirsung; the ventral bud contributes the small portion drained by the "duct of Santorini".
The anatomy of the pancreatic ducts is variable. Don't worry about this unless you are a surgeon.
In a few % of autopsies, one or more pancreatic choristomas are found somewhere in the gut. These are confusing when found, but probably cause no problems.
An "annular pancreas" is wrapped around the duodenum, and gets blamed for obstruction.
{49140} annular pancreas; sideways, liver on left, pancreas extends across duodenum and gall bladder
The adult organ varies widely in size ("Big Robbins"'s 60-140 gm seems reasonable). Anatomists talk about "head, body, and tail", which are visible with a little imagination; you may even hear about a "neck".
THE PANCREAS IN SYSTEMIC DISEASE
You remember that cystic fibrosis is a dread, common disease that causes atrophy of the pancreatic acini and malabsorption. It's now clear that some mutant CFTR homozygotes have normal sweat chloride studies and only chronic pancreatitis (Gut 52-S2: S-31, 2003; NEJM 339: 645, 1998; Dig. Dis. Sci. 45: 2007, 2000; Chest 126: 1215, 2004).
{00044} cystic fibrosis; trichrome stain,
showing dilated ducts plugged with good, and absent acini
{20204} hemochromatosis; Prussian Blue stain
{24505} hemochromatosis, gross, one's a Prussian blue
{24506} hemochromatosis, rusty color
{38848} hemochromatosis, rusty color. Scar tissue is white.
This is defined as inflammation of the exocrine pancreas with damage to the acinar cells.
While "Big Robbins" distinguishes "acute interstitial-edematous" and "acute hemorrhagic-necrotizing",
these probably describe varying degrees of severity of the same underlying process. "Big Robbins" is
also simply wrong to say acute pancreatitis is "by no means common"; it is one of the most common
severe intra-abdominal processes in adults.
Whatever the etiology in a specific case, pancreatitis is perpetuated by release of activated digestive
enzymes into the organ and surrounding tissues. Trypsin is presently credited with activating the other
enzymes, as well as kallikrein (remember that?). Whatever releases trypsinogen from cells and activates
it must be the cause of pancreatitis.
Chronic pancreatitis is a misnomer for pancreatic insufficiency and/or pain that probably is caused
by a previous episode of acute pancreatitis.
Because of its nature, any injury to the acinar cells will release digestive enzymes (trypsin, elastases,
other proteases, amylases, lipases, phospholipase) and result in more extensive damage.
"Idiopathic hereditary pancreatitis" often results from
a mutant trypsinogen gene (PASS1, Lancet 354: 42, 1999; Gut 44: 259, 1999).
Also common is a mutated pancreatic protease inhibitor (SPINK1 antitrypsin:
Gut 53: 723, 2004; Gut 50: 687, 2002).
The diagnosis of pancreatitis is generally made on the basis of finding damage to the pancreatic acinar
cells, i.e., by finding an elevated blood amylase and/or lipase.
Pitfall: About 1% of humankind has "macroamylasemia", a "large amylase" molecule (i.e., one bound
to an autoantibody) that is cleared abnormally slowly via the kidney (i.e., the urinary
amylase is not increased even though the serum amylase may be extremely high). This accounts for many
mistaken diagnoses of pancreatitis by the unwary.
What damages the exocrine pancreas? The list in "Big Robbins" is worth remembering:
Very heavy drinking (not exactly the same thing as "alcoholism"...) is the most important cause of
pancreatitis in the U.S., and many extreme alcoholic debauches end this way, and it is likely to be
severe.
How alcoholic excesses cause pancreatitis is remains mysterious.
Old thinking focused on malfunctions of the sphincter of Oddi, allowing reflux of enterokinase and/or
other noxious stuff; and/or increased production of secretin in the gut.
Experimental choline deficiency scrambles transport of proenzymes within acinar cells; they end up
activating one another. Binge drinkers don't keep a good, balanced diet....
* Your lecturer predicts that when the real cause of alcoholic pancreatitis is found, it will be a
physicochemical change in the subcellular membranes that causes intra-cellular activation of
pro-enzymes.
Cholelithiasis accounts for a large minority of cases of pancreatitis.
Gallstones are thought to cause pancreatitis:
(1) by lodging in the common bile duct and blocking the pancreatic duct, producing back-pressure
which pushes enzymes from the duct back into the interstitium, and/or;
(2) by damaging the sphincter of Oddi, allowing backwash of enterokinase and/or cell poisons such as
lysolecithin into the pancreas.
In the absence of a really solid stone, biliary sludge (i.e., many tiny particles in suspension, preventing
flow of bile) has probably been a long-unrecognized cause of pancreatitis (NEJM 326: 589, 1992).
Indeed, the smaller the gallstone, the more pancreatitis risk: Arch. Int.
Med. 157: 1674, 1997.
Less common are:
Trauma (i.e., a steering-wheel injury, or a poke at surgery)
Something bad in the general area, notably a perforated gastric ulcer
Viruses (i.e., mumps, cytomegalovirus; herpes simplex Arch. Path. Lab. Med. 127: 231, 2003)
Worms (clonorchis, ascaris)
Ischemia (i.e., horrible atherosclerosis)
Vasculitis (i.e., rickettsial disease, polyarteritis nodosa, etc.) Drugs (most notably big doses of
morphine)
Hyperlipidemia (types I and V; figure that one out!)
Hypercalcemia (no one knows why; * possibly it activates trypsinogen Gastroent. 109: 239, 1995; or
* prevents the zymogens from leaving the acinar cells, eventually getting them digested Am. J. Surg. 169:
167, 1995)
Certain CFTR carriers: NEJM 339: 645 & 653, 1998.
Following cardiopulmonary bypass (from calcium chloride in the resuscitation solution? NEJM 325:
382, 1991)
Drugs. Glucocorticoids, ddI, pentamidine,
and azathioprine (Am. J. Gastro. 98: 1305, 2003) are famous.
Uremia (i.e., renal failure) Heredity (see below)
* Pancreas divisum (i.e., failure of the two buds to fuse, with Santorini's duct awkwardly draining the
body and tail) affects 3-10% of people and is touted as an additional "cause of pancreatitis"
because most of the pancreas drains through the smaller duct.
If this really causes pancreatitis, it is uncommon (Int. J. Pancr. 5: 317, 1989).
Patients present with abdominal and/or back pain, fever and shock; the latter are probably due to
contamination of the bloodstream by foul products of autodigestion.
In addition to hyperamylasemia, patients may have obstructive jaundice (understandable!),
hypocalcemia ("from all that fat necrosis calcifying", and/or some substance that depresses the
parathyroid glands), and/or elevated blood glucose.
They may develop duodenal obstruction, ARDS, and/or acute tubular necrosis of the kidney (more
about the last one under "Kidney").
* Future pathologists: A few days after death, the autolyzing
pancreas, though normal in life, may look intensely
hemorrhagic. Don't be fooled.
Microscopic views will clarify the issue.
At surgery ("close him back up") or autopsy ("a familiar finding"), acute pancreatitis is unmistakable.
In the acute process, the pancreas undergoes proteolysis (proteases) and lipolysis (lipase), eventually
with hemorrhage (elastases damage vessels). Not surprisingly, this produces considerable acute
inflammation (neutrophils, edema, and so forth).
Ascites in these patients is ugly brown and often has globules of fat floating on its surface, like on
chicken soup. Of course, it is loaded with amylase.
You are already familiar with fat necrosis ("saponification", etc.) The dead cells may fill with
amorphous debris and/or calcify (calcium complexes with free fatty acids) heavily enough to have
caused hypocalcemia. Grossly, fat necrosis looks and feels much like chalk.
Even years later, spots of old fat necrosis may stud the omentum (though the polys are gone after the
acute phase). Finding a few flecks of fat necrosis at autopsy is of no significance, and could be agonal
(i.e., the result of the ischemic death-throes of the pancreas). Lots of fat necrosis is very suspicious for previous
pancreatic injury.
{08339} enzymatic fat necrosis, gross; it is the
chalky granular stuff. The background is hemorrhagic pancreatitis.
Oddly, these patients can also have fat necrosis at sites remote from the pancreas, i.e., under the skin.
The mix of processes gives the acute case a variegated pattern of blacks, browns, reds, yellows, and
off-whites.
After the acute phase is over, liquefied areas may be surrounded by fibrous tissue, producing a
pseudocyst ("pseudo" because there is no epithelial lining). If they become infected while forming, a
pancreatic abscess results. The biggest pseudocysts replace the lesser sac, which was autodigested.
{49233} pseudocyst; spleen at right. Hollow and filled
with fluid.
It is worth remembering that the duodenum can become obstructed in pancreatitis.
Future radiologists: Look for a paralyzed segment of bowel ("sentinel loop") near the sick pancreas.
Chronic pancreatitis probably is the result of scarring from one or more episodes of pancreatitis, which
have often not been obvious clinically.
It is usually seen in chronic problem drinkers, as a pain syndrome associated with nerve involvement.
There is loss of acinar cells (later, even the islets are gone), atrophy of some of the remaining cells, and
dense fibrous tissue; scar contraction is likely to dilate the ducts.
Grossly, this produces a small, firm, white pancreas.
* Squamous metaplasia of the ducts
may occur, perhaps because the cells are seeing so much more irritating material.
Pathologists also look for calcifications ("chronic calcifying pancreatitis"). These may be either (1)
calcified fat necrosis, or (2) "pancreatic calculi", dystrophic-calcified lumps of protein (* error in
Walters: it's not lactoferrin) in the pancreatic duct. The protein is assumed to be some digestive thing,
though it has eluded precise characterization.
* Many people think that "protein plugs", their production somehow
stimulated by alcohol excess, "cause" acute
alcoholic pancreatitis. This is unlikely, since the histology of chronic alcoholic pancreatitis is different
from the obstructive lesion (Am. J. Gastroent. 85: 271, 1990). See below.
The scarring may produced unorganized-appearing glands, which the unwary pathologist may mistake
for cancer.
Future pathologists: Sometimes it is very hard to tell well-differentiated
adenocarcinoma from scarring
here. Clues to cancer included mitotic figures, necrotic debris, incomplete lumens, and widely-variable
nuclear sizes (4:1 or more); today's pathologist may also stain for k-ras mutations (Am. J. Clin. Path. 105: 321, 1996),
and other genes (p53, more arcane ones Am. J. Clin. Path. 117:
755, 2002; even on fine-needle aspirates).
{49234} chronic pancreatitis; pale white is scar
In the late stages, patients can expect to have malabsorption (steatorrhea, weight loss), and perhaps a
pseudocyst. "Big Robbins" does not mention the worst problem that many of these people have --
chronic severe pain from involvement of the sensory nerves around the celiac plexus.
As you must have noticed, there is little justification for calling this "chronic pancreatitis", except that
it may be chronically painful. This misnomer was canonized in the Marseilles-Rome criteria of
1988
(Scand. J. Gastroent. 24: 641, 1989).
Chronic obstructive pancreatitis, a slightly different entity from the above, follows obstruction of the
pancreatic duct (gallstone, surgeon's mishap). In this situation, there is selective atrophy of acini around
the head of the pancreas. Surgeons can repair the ampulla if that is the problem.
* Exactly how the cells die remains obscure; there are conflicting animal models (apoptosis vs.
inflammation/necrosis): Gastroenterology 110: 875, 1996.
* The most interesting work in chronic pancreatitis recently is
the discovery that antioxidants seem to help a lot: Am. J. Gastro. 91:
1558, 1996, others.
Future pathologists: Here's how to distinguish these two entities (after World. J. Surg. 14: 2, 1990):
NON-MALIGNANT MASSES
* Cysts of the pancreas are rare. They are seen (in a minority of cases) in two probable
anti-oncogene-deletion syndromes (Von Hippel-Lindau, adult polycystic kidney disease).
Pancreatic pseudocysts (see above) are common after acute pancreatitis from alcoholism or trauma, or
from any other cause.
Benign tumors of the pancreas are uncommon. They are almost always some variant of adenoma.
Cystadenomas are the most common. Leave it to the pathologist
to decide whether a particular tumor has malignant potential Cancer 71: 82, 1993.
* Intraductal papillary neoplasms: Cancer 72: 689, 1993 (can express both exocrine and endocrine
markers, which is not surprising, since both come from ducts); Ann. Surg. 223: 141, 1996.
Dysplastic ones can probably turn into cancer.
{49238} cystadenoma of the pancreas; has been
cut in half and opened; spleen at right
CANCER OF THE PANCREAS ("cank of the pank", "the dismal disease",
etc.; Lancet 363:1049, 2004; Disease-A-Month 50: 545, 2004)
This dread cancer accounts for about 5% of U.S. cancer deaths; the incidence has tripled in the past 50
years "because of smoking and chemicals" (I wonder).
* Famous victims in recent memory include Billy Carter, Jack Benny, Dizzy Gillespie, Rex Harrison,
and Michael Landon.
Virtually all cancers of the pancreas are adenocarcinomas arising from the ducts.
Like adenocarcinomas anywhere else, you can spot them because they make little glands and/or are
secretory- product (i.e., mucin)-positive.
{08851} adenocarcinoma of pancreas;
no normal pancreas on the slide; some glands are more anaplastic than
others;
Future pathologists and surgeons: Cancer of the pancreas and chronic pancreatitis are hard to tell apart.
A certain percentage of false-positive diagnoses of cancer of the pancreas, and a certain number of
Whipple procedures for those without cancer of the pancreas, is acceptable: Br. J. Surg. 81: 585, 1994.
Future pathologists only: The in-situ evolution of pancreatic cancer has been well-studied. See Arch.
Path. Lab. Med. 118: 227, 1994.
* For some reason, it is not uncommon to see real osteoclasts and even osseous metaplasia here (J. Clin.
Path. 47: 372, 1994; Arch. Path. Lab. Med. 120: 306, 1996); these are still carcinomas.
"Big Robbins" links it to the notable carcinogens
naphthylamine and benzidine, and the hoopla over nitrosamines in food was related to their link to
cancer of the pancreas in experimental animals.
* Questionable risk factors include alcohol consumption, high-fat diet ("cholecystokinin must be a
promoter"; tough to believe if you think pancreatic acinar cells don't ordinarily divide), coffee drinking,
obesity, and pernicious anemia. All of these are now pretty much discredited (see for example Cancer
67: 2664, 1991; the coffee crock discredited Cancer Epidem. 10: 429, 2001, several
others uniformly negative).
Anti-oncogene deletion syndromes placing people at excess risk for pancreatic cancer include
BRCA2 (breast-and-ovary), the Lynch hereditary nonpolyposis coli cancer syndromes
(hMSH2, hMLH1), Peutz-Jegher's (STK1/LKB1; Big Robbin's claim of a x130 increased risk
can't be right), and the p16/CDKN2A pancreatic cancer/melanoma syndrome (NEJM 350: 2623, 2004).
Hereditary pancreatitis in particular gives at least a 40% risk of getting
pancreatic cancer (Med. Clin. N.A. 84: 719, 2000).
* Concerns about exposure to particular pesticides keep coming up,
but the whole business remains very soft -- the ones that "mutate k-ras
in lab animals" aren't the ones that "are associated with a 5x increased
risk of cancer of the pancreas in workers", etc., etc. To become
confused, see Env. Health Perspect. 111: 724, 2003; Lancet 354:
2125, 1999.
Whatever the environmental "cause", most (or maybe all) cancers of the exocrine pancreas have mutated
k-ras at hot-spot codon 12. This can be detected on fine-needle aspirate material, and by PCR
in pancreatic fluid (Cancer 73: 1589, 1994; Am. J. Path. 144: 889, 1994) or stool (ooh, Cancer Res. 54:
3568, 1994) or smears (Am. J. Clin. Path. 105: 257 & 321, 1996).
Smoking seems to cause this mutation (Cancer 85: 326, 1999).
The distribution of cancers in "Big Robbins" is reasonable:
60% head
Patients come in with back pain (why?), jaundice, weight loss, GI upsets, depression (very typical, and
poorly understood), and/or migratory thrombophlebitis ("Trousseau's other sign"; the mechanism of the
distinctive paraneoplastic problem is unknown).
The size of the cancer depends on the stage at which it is detected. Those in the head may be found
early because they produce jaundice. Those in the body and tail will be detected late.
There's a serum tumor marker, CA-19-9 (Gut 35: 707, 1994, many others). * Another, for cyst fluid:
CA-72-4 (Ann. Surg. 219: 131, 1994).
Future surgeons: Courvoisier's law states that a distended gall bladder in a patient with obstructive
jaundice means cancer (pancreas, common bile duct). Obstruction due to a gallstone in the common
bile duct will not result in a distended gallbladder, because the gallbladder would be heavily scarred-up
from years of cholelithiasis. This works most of the time, though you would never rely on it.
Gung-ho surgeons may try to resect a tumor in the head of the pancreas ("Whipple procedure"; * for the
pylorus-sparing technique see J. Am. Col. Surg. 178: 443, 1994; for the Hopkins study, which hails
11 cures out of 201 surgeries as an enormous improvement, see Ann. Surg. 221: 721, 1995. Desperate
diseases require desperate remedies).
The overall 5-year survival rate in cancer of the pancreas is about 1%; typical survival is about six
months (Gut 31: 494, 1990; J. Am. Col. Surg. 179: 38, 1994).
These patients often have diabetes, and the cause is insulin resistance. This now appears to be due to
massive production of amylin (IAPP; NEJM 330: 313, 1994; Gastroenterology 114:
130, 1998); probably the amylin is produced by the islets
in response to one or more factors produced
by the tumor itself (J. Clin. Endo. Metab. 85: 1232, 2000). This probably
explains the well-known "link" between pancreatic cancer and diabetes, and it now appears that only
new-onset (i.e., less than three years) diabetes is a "risk factor" (NEJM 331: 81, 1994).
* A recent British euthanasia case involved intractable pain from cancer of the pancreas: Lancet 335:
719, 1990 ("not guilty"; in my opinion this is a triumph of humanity and common sense; you may
disagree).
Adenocarcinoma of the pancreas typically metastasizes to lymphatics, and blood-borne metastases to
the liver are typically massive.
* Acinar carcinoma of the pancreas is a rare PAS-positive tumor,
usually of younger people. It
presents stippled cells that stain for amylase, lipase,
and chymotrypsin, and often
elaborates lipase into the blood (which may
produce subcutaneous fat necrosis!) Molecular genetics: Am. J. Path. 160: 953, 2002.
CLASSIFYING DIABETES MELLITUS AND RELATED CONDITIONS
Diabetes mellitus (MELL-uh-tuss, please) is "a chronic disorder of carbohydrate, fat, and protein
metabolism characterized in its fully expressed clinical form by an absolute or relative insulin
deficiency, fasting hyperglycemia, glycosuria, and a striking tendency toward the development of
atherosclerosis, microangiopathy, nephropathy, and neuropathy" (old Big Robbins).
Diabetes is our commonest serious metabolic disease, affecting maybe 5% of the population.
On the average, it
takes 15 years off the patient's life (JAMA 285: 628, 2001) and accounts
for a tremendous amount health care expenses.
You will
need to know the terminology (which is often not used correctly):
Diabetes mellitus ("overt diabetes", "manifest diabetes", etc.): the patient has...
There's talk today about distinguishing
"impaired fasting glucose" (IFG, i.e., 110-125 mg/dL)
and
"impaired glucose tolerance" (IGT, i.e., 121-179 mg/dL at the two-hour mark).
See Arch. Int. Med. 161: 397, 2001.
Type I diabetes and Type II diabetes (below) are sometimes called "primary diabetes", since they seem
to be genetic diseases in their own right.
Secondary diabetes is said to exist when the metabolic disturbances are the result of some other
identifiable illness, injury, molecular abnormality, etc., etc.
Impaired glucose tolerance ("glucose intolerance", "subclinical diabetes", "asymptomatic diabetes",
"chemical diabetes", "latent diabetes"): fasting blood sugar is normal,
but a glucose tolerance test is
abnormal. Current recommendations are NOT go looking for this:
Am. J. Med. 105(1A): 15S, 1998.
Gestational diabetes mellitus: diabetes mellitus first appearing during pregnancy, and perhaps
disappearing when the pregnancy ends.
"Previous Abnormality of Glucose Tolerance" ("prediabetes", "latent diabetes"): the patient once had
measurable glucose intolerance (as, when she was pregnant), but is chemically normal now (but may
be at risk for future diabetes mellitus, depending on the circumstances).
"Potential Abnormality of Glucose Tolerance" ("prediabetes"): the monozygotic twin of a type II
diabetic, or (less justifiably) someone else with a strong family history.
Not diabetes: Glucose intolerance only under some obvious physiologic stress (myocardial infarction,
pneumonia, severe burns, terror of venipuncture, etc.) Mostly an epinephrine effect;
probably cortisol contributes as well.
Type II: defects in insulin secretion, and/or a relative lack of
insulin, and/or insulin resistance;
Type III: damage to the whole pancreas (old pancreatitis,
cystic fibrosis -- diabetes from CF is uncommon but happens J. Ped. 142: 97, 2003) and autosomal dominant genetic
syndromes (I'm clueless
as to why these are lumped together);
Type IV: Gestational diabetes
PRIMARY DIABETES TYPE I ("juvenile onset", "labile",
"ketoacidosis-prone", "insulin-dependent"): 10% of diabetics.
One person in 300 in the U.S. gets this kind of diabetes (rates vary considerably from nation to nation;
* rates are higher at higher latitudes).
Typical case: A child (average age twelve years, but we now know you can get the disease at any age) presents with
polyuria, polydipsia, and polyphagia of relatively sudden onset. The child is found to have very high
blood glucose levels causing osmotic diuresis.
Before the era of injectable insulin, diabetic ketoacidosis (DKA) and death followed in short order.
You remember the pathophysiology of ketoacidosis from your physiology course. Future clinicians:
Ketoacids impart the familiar "rotten apples" sweetness to these patients' breath.
Today, the child looks forward to a period of fairly good health while taking injectable insulin, checking
blood glucose several times a day with chemical strips and a reflectance meter.
After 10-15 years, unless control is good,
the diabetic starts to suffer with
infections, eye problems, peripheral neuropathy, gangrene of the lower extremities,
kidney disease, stroke, and coronary atherosclerosis.
Historically, death
usually came about forty years after onset
as the result of a myocardial infarction. By this time, 50% of patients
had lost their kidneys, and nearly as many were blind, stroked out, legless, and/or in chronic pain from
neuropathy. A well-treated, compliant diabetic typically does better today.
The essential lesion in type I diabetes is a severe absolute lack of insulin.
* Only half of patients have any evidence of insulin production (measure C-peptide in serum).
Insulin deficiency and hyperglycemia explain the presentation but do not explain the later complications
of the disease.
"Type I diabetes is a genetically programmed, chronic autoimmune disease"
(NEJM 314: 1360, 1986,
an early review; update Nature 351: 519, 1991), with the acute-symptomatic phase sometimes
triggered by an acute viral illness. In other words, the etiology is kind-of-complex.
Genetic factors:
Siblings of those with Type I diabetes are at increased risk (25x).
Identical twins of those with Type I diabetes have a 50% chance of eventually getting it also.
Type I diabetes is strongly associated with HLA-related antigens DR3 and DR4. (* If one has the
misfortune to have both, it's even worse.... The former association with some HLA-B antigens was due
to their linkage to DR3 and DR4; and currently, it appears that the also-linked DQ is the closest
important site.)
* As is so common when the immune system attacks gland parenchyma,
the beta cells of these patients express HLA class II
histocompatibility antigens. No one knows whether this is cause or effect.
* The molecular defect that permits type I diabetes to occur seems to be homozygous absence of aspartic
acid in position 57 of the HLA class II DQ chain (Nature 329: 599, 1987; Nature 333: 710, 1988), at
least in the U.S. Update on HLA links: J. Clin. Endo. Metab. 89: 4037, 2004;
they vary tremendously from nation to nation: J. Clin. Endo. Metab. 90:
5104, 2005.
The famous locus IDDM1, where certain polymorphisms give
a risk for type I diabetes, is a component of the HLA system (Diabetes 50:
1200, 2001).
The gene IDDM2 ("implicated in diabetes melitus") is a complicated,
highly variable tandem repeat adjacent to the real insulin gene.
Two variants are strongly linked to type I diabetes (update Diabetes 53:
1884, 2004).
The animal model of autoimmune diabetes is the non-obese diabetic mouse, which gets that way
because of genes at three (or more) loci (Nature 353: 260, 1991; J. Imm. 152: 204, 1994).
Update J. Immuno. 169: 6617, 2002; to date; the exact
reasons
for the famous mouse's diabetes remain elusive.
* The BB (formerly BB/W) rat is a strain discovered in 1977. These rats have autoimmune insulitis, and the majority
develop acute-onset type I diabetes. They helped us find
the IDDM1 and IDDM2 loci
(Acta. Diabet. 35: 109, 1998).
Autoimmune factors:
Several types of IgG anti-beta-cell antibodies occur. One or more is present in the
vast majority of type I diabetics the acute
phase (contrast 0.5% in healthy people). It is now quite clear
that they are etiologic, and that they are usually present before age 2
in children destined to get type I diabetes (Ann. Int. Med. 140:
882, 2004).
Their specificities include
anti-glutamic acid decarboxylase (Nature 347: 151, 1990; NEJM 322: 1555, 1990; Lancet 341: 1378 &
1383, 1993; diabetogenic epitope Lancet 343: 1607, 1994; true both of NOD mice and people: Nature
366: 69 & 72, 1993).
There is also cell-mediated immunity directed against beta cells in most patients who have been studied.
Again, the autoantigen is glutamic acid decarboxylase.
Update Nature 391: 177, 1998.
There was generally a dense lymphocytic infiltrate in the islets of patients dying in the acute phase (rare
nowadays).
* Maybe 1 in 5 of these people ends up with another autoimmune glandular disease (autoimmune Addison's
disease, Hasmimoto's autoimmune thyroiditis, Grave's disease of the thyroid). Likewise, plenty of
people, with or without other autoantibodies, have anti-islet cell antibodies but never go on to develop
autoimmune diabetes.
The claim from the early 1990's that cow's milk is the trigger
for autoimmune diabetes flopped
(JAMA 276: 609 & 647, 1996, NEJM 329:
1853, 1993, J. Clin. Endo. 87: 3192, 2002;
the activists keep the business going but there now seems
to be a consensus there is nothing real here: Eur. J. Clin. Nutr. 59:
623, 2005; Nutrition 21: 474, 2005).
* The non-obese diabetic mouse does get some protection from drinking mother's
milk instead of cow's milk.
The experimentalists speculat at length about how perhaps this is because mother's milk
contains insulin and/or other peptides
to which the gut lymphocytes need to become tolerant (Diabetes 48:
1501, 1999). But think -- the experiment requires taking the experimental
mice away from their mothers. This must have many far-reaching
effects beyond just the exposure to cow's milk.
A few groups are "curing" mice of type I diabetes using immune manipulation
(Freund's adjuvants, etc., etc.) Update J. Clin. Inv. 108: 63, 2001.
Viral factors: Clinically, Type I diabetes often follows a viral illness.
Worth knowing: Kilham rat parvovirus infection produces type I autoimmune diabetes in
diabetes-resistant rats (Diabetes 45: 557, 1996; J. Immuno. 165: 2866, 2000).
This is now a robust finding (J. Imm. 173: 137, 2004).
* Retrovirus IDDMK(1,2)22 remains controversial as a cause of type I diabetes. Some folks
don't find it at all (Diabetes 48: 209 & 219, 1999); others
find soft data suggesting a link (J. Hum. Genet. 46: 712, 2001).
A Coxsackie B4 virus from the pancreas of a patient dying shortly after the onset of the illness
destroys the beta cells of NOD (non-obese diabetic) mice; it's now clear that the virus causes a chronic infection of these
islands
(J. Inf. Dis. 171: 1131, 1995). Since this article, Coxsackie CB4
has been found commonly as a recent infection in kids coming down with
diabetes.
The mechanism of Coxsackie B4 induction of diabetes now seems clear --
the NOD mouse has lots of autoreactive-but-unactivated T-cells,
Coxsackie B4 produces a mild infection of the beta cells, and bystander T-cells
are activated ("bystander activation"). Happens in mice and maybe
in kids.
* There have been outbreaks of type I diabetes in which the children
were found to produce enterovirus mRNA; these patients do not show the usual
HLA antigens cited as placing a person at risk, and perhaps this is
a distinct subcategory with a specific viral cause (J. Inf. Dis. 187:
1562, 2003).
Overwhelming infections with mumps or cytomegalovirus also
have been implicated in rare cases of "type I diabetes". The pancreas
can be destroyed by congenital rubella.
* A huge search for
the "insulitis virus" in humans
using molecular probes found nothing: JAMA 257: 1145, 1987.
What does all this mean? In most cases of type I diabetes, it is hypothesized that a viral infection
triggers autoimmune destruction of the beta cells in genetically-predisposed individuals.
However, in most of these children, there have been progressive abnormalities of glucose metabolism
in these patients long before the onset of illness (Br. Med. J. 294: 5, 1987).
Some stress ("maybe the virus") apparently causes decompensation at the "time of onset". Following
recovery from the first episode of ketoacidosis, the "honeymoon period" begins, when control is easy
for several years. (* Patients continue to produce some of their own insulin -- i.e., there is C-peptide in
their blood -- during the "honeymoon".)
* No one knows why type I diabetes is becoming more common,
but there's no question that this is happening.
Studies that suggest a major, unknown environmental factor
(Lancet 364: 1699, 2004) make me think we are dealing with a viral trigger.
PRIMARY DIABETES TYPE II ("adult onset", "stable",
"ketosis-resistant", "non-insulin-dependent"): 90% of diabetics.
Typical case:
An overweight adult (most over age forty) is discovered on routine screening to have elevated fasting
glucose or glycosuria.
In other cases, the diabetes is discovered during evaluation of impotence, pain, eye trouble, stroke, foot
trouble, bad infection, or coronary disease.
Some patients have their diabetic predisposition unmasked by pregnancy. Such women get better after
the pregnancy, but are at greater risk for eventually developing type II diabetes.
Before the era of injectable insulin, nothing much was done for type II diabetics, even if the disease was
detected. The patients got complications and had shorter life spans.
Today, the adult looks forward to dieting, doing aerobic exercise,
and possibly getting treated with insulin or "diabetes
pills", probably getting an ACE inhibitor, and maybe a statin for lipid control.
Complications will occur as in Type I diabetes, depending on how
well the patient is able to manage the hyperglycemia.
Death will probably be due to a myocardial infarct.
Type II diabetes is a polygenic disorder, with its expression modified by a person's exercise habits and
amount of bodyfat.
Identical twins have nearly 100% concordance for Type II diabetes.
There are no good HLA associations or phenomena pointing to autoimmunity.
A subtype of Type II diabetes that can present in young people (maturity-onset diabetes of the
young, MODY) is an autosomal dominant with 90% penetrance, and several loci.
See below.
MODY accounts for about 10% of diabetics in some communities,
and less-severe alleles of the genes are of course implicated in common type II diabetes.
The defect is usually in the glucokinase gene (Nature 356: 721, 1992; mechanisms Lancet 340: 444,
1990; diagnosis Lancet 345: 1313, 1995;
pathophysiology Diabetes 46: 204, 1997;
this enzyme, as you remember, is the key link in the
signalling system by which beta cells monitor blood glucose).
There are a few other MODY genes too,
all in the insulin-release system (Proc. Nat. Acad. Sci. 94:
13209, 1997; Diabetes 47: 1459, 1998;
Diabete 52: 872, 2003). Update on genes
Diabetes 53:
1894, 2004.
A single major genetic defect at a type II diabetes locus, and/or several
minor defects at several of the loci, seems to be the underlying
cause of type II diabetes.
The insulin resistance genes to date, and the percentage of type II diabetics with each (Nature 373: 384,
1995, also Am. J. Clin. Path. 105: 149, 1996):
1%... mitochondrial DNA syndromes (often goes with deafness; Ann. Int. Med. 134:
721, 2001; many others)
?%... the mitochondrial uncoupling proteins (Diabetes
47: 1528, 1998; Diabetes 53: 1905, 2004).
1%... glucokinase
1%... insulin itself
1%... insulin receptor (* the severe form is "leprechaunism", a progeria: Biochim. Biophys. Acta.1402: 86, 1998)
15%... insulin receptor substrate (IRS-1, it's very complicated and results are mixed: Diabetes 52: 1544, 2003; J. Clin.Inv. 114: 908, 2004)
1%... GLUT4, the glucose-through-the-membrane transporter
?%... adiponectin, released from adipocytes, causes liver and muscle to burn triglyceride and be more insulin-sensitive (Nat. Med. 7 887, 2001;
Nat. Med. 10: 452 & 524, 2004; Diabetes 53: 1150, 2004).
?... adiponectin receptors 1 and 2 (Diabetes 53: 2132, 2004; Diabetes 54: 2245, 2005)
?%... hepatocyte nuclear factor alpha (HNF-1alpha/TCF1, causes MODY3; risk for classic type II; Diabetes 53: 2122, 2004; Diabetes 54: 2336, 2005; Diabetes 53: 1141 & 3002, 2004)
?%... ICAM-1 (Lancet 362: 1723, 2003)
?%... calpain 10 (J. Clin. Endo. Metab. 87: 2606, 2002)
?%... beta adrenergic receptors (gives the munchies / obesity and diabetes: NEJM 333: 382, 1995; Clin. Endo. 59: 476, 2003).
?%... leptin (must be rare in humans, though of course in mice it's famous)
?%... phosphoenolpyruvate carboxykinase (J. Clin. Endo. Metab. 89: 898, 2004.
?%... Sulfonylurea receptor (Lancet 361: 22, 2003).
?%... HIF-1alpha (induces VEGF, J. Clin. Endo. Metab. 90: 5841, 2005).
?%... alpha2-Heremans-Schmid glycoprotein (Diabetes 54: 2477, 2005)
?%... mitochondrial fat-burning systems (no gene yet; NEJM 350: 664, 2004);
?%... mitochondrial leucyl tRNA synthetase / LARS2 (Diabetes 54: 1892, 2005)
?%... sterol regulatory element binding protein (SREBP)-1 (Diabetes 53:
842 & 2153, 2004
?%... K(ATP) channels in muscle (?!; Diabetes 54: 1592, 2005)
* The lipoatrophic diabetic mouse has zero bodyfat and extreme
insulin resistance with diabetes. A gene product awaits discovery
and characterization
(Diabetes 51: 2113, 2002). This is supposed
to be a model for both a few human genetic syndromes
and the lipoatrophy of HIV patients on protease inhibitors
(Ann. Int. Med. 133: 304, 2000).
Type II diabetes is now rampant in the
third world and many of our own First American peoples.
Until recently, the tendency was to blame the western diet
("the poor nations have been coca-colonized": (Nature 357: 362, 1992).
I have always taught that the real reason is that the world's poor
are much better-fed than in the past, and most no longer lead lives of
constant hard physical labor. Stay tuned.
Of course, there has been stronger natural selection against diabetes
in countries like the U.S. and Western Europe
that have been well-fed for centuries.
And in societies with episodes of famine, there is a strong selection bias for type II
diabetic body chemistry (i.e., a tendency to hang onto carbohydrate calories), and little chance to express
the phenotype.
Whether or not it's related, unborn children exposed to famine have a much stronger
tendency to develop type II diabetes when they grow up: Lancet
351: 173, 1998.
* The Pima Indians present a special problem; their rate of diabetes
is extremely high with a host of different genetic mutations for insulin
resistance (update Diabetes 53: 1181, 2004).
By age 65, the following percentages of U.S. ethnic groups have diabetes:
Hispanics 33%
The pathophysiology of type II diabetes is fairly well understood.
In type II diabetes, basal insulin secretion is generally normal. In response to glucose administration,
insulin secretion may be abnormally low, normal (rare), abnormally high, or delayed ("too much, too
late").
Most Type II diabetics have insulin resistance in both liver and skeletal muscle, and this appears to be
the key lesion. In addition, however, there is almost always some evidence of beta cell dysfunction.
The liver continues to make and put out glucose (gluconeogenesis) when blood sugar is high, and fails
to take up orally-administered glucose. The skeletal muscles fail to take up glucose in response to
insulin.
The amount of insulin resistance is modified
by obesity and physical conditioning.
There's also the baffling combination of
gut polypeptides, prostaglandins, beta-endorphins, etc., etc....
You already know the metabolic syndrome / metabolic syndrome X (truncal
obesity, insulin resistance, dyslipidemia). The cause remains obscure.
The most recent suspect is resistin, produced (at least in mice) by adipocytes,
and able to render muscle and liver resistant to the effects of insulin
(NEJM 345: 1345, 2001). It's produced especially well by the abdominal
and omental adipocytes; this may explain the special risk of "central obesity".
* Liposuction completely fails to alter the metabolic
abnormalities caused by obesity (NEJM 350: 2549, 2004).
Two other players in the complex business
of insulin resistance is a pair of little-known hormones, amylin
("islet-amyloid polypeptide", "IAPP", pumped out of beta cells along with insulin) and * calcitonin-gene
related polypeptide (CGRP, h-CGRP, from nerve and gut), both acting on skeletal muscle to increase
its resistance to insulin (PNAS 88: 7713, 1988). * They act on the same receptor, which is not present
in fat or parenchymal cells (Diabetes 40: 395, 1991; Diabetes 40(S1): #267, #255, several others,
1991).
Amylin has been reported to be greatly increased in the serum of some type II diabetics. Excreted
through the kidneys, it also might account for some of the insulin resistance in renal failure. See
Diabetes Care 39(S1): A111-A113, 1990.
More recently, many workers have concluded that neither hormone is present in sufficient quantities to
exert an important physiological effect in asymptomatic or diabetic humans (Diabetes 40: 305 & 310, 1991).
Nevertheless, the hormones have been conserved over mammalian evolution for some reason, and
assays and preparations are poorly-standardized (late 1990). Amylin update Lancet 341: 1249, 1993.
Most recently, genetically scrambled mice who overexpress amylin do get hyperglycemic, with a
syndrome much like human type II diabetes (Proc. Nat. Acad. Sci. 93: 3492, 1996). Definitely stay
tuned.
I predict that when the underlying cause of type II diabetes
(i.e., simultaneous insulin resistance and aberrant insulin production)
is worked out, it will prove to be primarily a mitochondriopathy. Stay tuned.
The "Somogyi phenomenon" is a rebound hyperglycemia from all the stress hormones
that pour out when the blood glucose drops too low from too much insulin.
If a diabetic is hungry, gaining weight, and feeling crummy, consider reducing
the insulin levels.
The "dawn phenomenon, i.e., hyperglycemia and insulin resistance
in the morning without previous
"Somogyi"
hypoglycemia, is due to the high output of hGH during while you're finishing up
your sleep
in the morning.
* WARNING: Many clinicians use the term "insulin resistance" to refer instead to hard-to-manage
diabetics of any type who require more than 200 units of insulin daily (a whopping dose). Many of
these patients ave antibodies against insulin, while others have severe type II diabetes or any of several
other problems. See NEJM 315: 212, 1987.
Hyperosmolar nonketotic diabetic coma (HNKK, HONK) is the usual cause of "diabetic coma" in Type
II diabetics (see Arch. Int. Med. 147: 499, 1987), though most of them never get it.
Classically, some acute stress (often the 'flu) increases the
demand on the Type II diabetic's struggling beta cells,
and the supply of insulin is exhausted. Plasma glucose levels suddenly go extremely high, causing
osmotic diuresis, electrolyte disturbances, and death.
Or the illness may simply cause dehydration, producing a vicious cycle with insulin
resistance, stress hormones, soaring glucose levels, and ongoing dehydration.
Ketoacidosis is uncommon in type II diabetes, but can occur.
SECONDARY DIABETES has many etiologies
Pancreatic diabetes: destruction of the islets by disease of the exocrine pancreas.
Causes: pancreatitis, carcinoma, hemochromatosis ("bronze diabetes" -- don't overlook this one!),
trauma, surgery, etc. etc.
Endocrine diabetes: glucose intolerance due to other endocrine disturbances
Causes: Cushing's syndrome (from any cause), acromegaly, amylin from pancreatic cancer, obesity (??),
stress, amylin production by cancer of the pancreas (see above), etc. etc. It would be logical to place
pregnancy here too, though it is officially classed elsewhere.
Some people put the one-gene insulin resistance syndromes here, which
makes less and less sense every year as these genes turn out to be alleles for
standard Type II diabetes.
Rarely, people make autoantibodies that block insulin receptors (South. Med. J. 92:
717, 1999). Update J. Clin. Endo. Metab. 89:
2222, 2004; contrary to popular belief, acanthosis nigricans in a young
diabetic (while commonly seen) does not imply antibodies to insulin receptors.
REMEMBER: Regardless of the cause of the prolonged hyperglycemia, we now know that the
complications in remote organs (arteries, eyes, kidneys, nerves) will be the same.
ANATOMIC PATHOLOGY OF DIABETES MELLITUS: These are usually the effects, rather than the
causes, of hyperglycemia.
DIABETIC BLOOD VESSEL DISEASE
LARGE VESSEL DISEASE ("macroangiopathy"): accelerated atherosclerosis
Diabetics have a variety of poorly-understood disturbances of lipid metabolism. Nonenzymatic
glycosylation of lipoproteins seems to be a problem, LDL's stick best to glycosylated collagen, etc., and
glycation products (when they bind to their special receptors in the intima)
cause the production of fibrous tissue.
The result is the rapid development of severe atherosclerosis, with strokes, gangrene of the lower
extremities, and myocardial infarcts taking their toll, often early in life. Of course, this is all much
worse if the diabetic also smokes cigarets.
Big news: Administering the soluble form of the glycation product receptor
seems to stop the accelerated atherosclerosis. Definitely stay tuned.
Nature 4: 1025, 1998.
{09378} diabetic gangrene
SMALL VESSEL DISEASE ("microangiopathy"): hyaline arteriolar sclerosis
This is a complex problem.
The basement membrane of the capillaries and the arterioles becomes much thicker ("hyaline arteriolar
sclerosis"). Its expansion eventually compromises the lumen of the vessels.
Not surprisingly, these vessels are relatively inelastic, and this is an early, important problem: Br. Med.
J. 312: 744, 1996.
Even if the lumen is not badly compromised and the wall isn't excessively stiff, the small vessels of
diabetics open and close chaotically, and proper tissue perfusion cannot be assured.
Additionally, the pericytes can proliferate (especially in the glomeruli, where pericytes are called
"mesangial cells") or die off (especially in the retina, where pericytes are called "mural cells"). This
causes trouble at both sites.
* Endothelial cells can also proliferate, narrowing the lumen further.
* Other factors that are cited are the over-sticky platelets of diabetics, increased blood viscosity,
increased RBC rigidity, and increased numbers of free radicals.
Microangiopathy augments the ischemia caused by atherosclerosis, which is why so many diabetics lose
legs. It may account for other problems also.
Yes! Tight diabetic control reduces and even reverses microangiopathy. See NEJM 309: 1546 & 1551,
1983, and many others since.
Most diabetics eventually become hypertensive. Nobody knows why, but inability to handle sodium
seems essential: Am. J. Med. Sci. 307(S1): S-53, 1994.
Many diabetics are greatly troubled by congestive heart failure as the disease progresses,
and perhaps nonenzymatic glycosylation of the heart muscle proteins itself is
part of the problem, since even if you control for other factors, poor glycemic control
correlates strongly with the development of CHF (Circulation 103 2668, 2001).
DIABETIC KIDNEY DISEASE ("diabetic nephropathy"; Disease-A-Month
44: 214, 1998; NEJM 341: 1127, 1999):
Renal failure causes much disability and death among type I diabetics; this is now the #1 single cause
of end-stage renal disease in the U.S. Type II diabetics generally die of something else before their
kidneys fail.
Renal vascular lesions
Arteriolar sclerosis of both afferent and efferent arterioles at the glomerular pole is highly characteristic
of diabetes. (The other diseases of renal arterioles, notably common-type high blood pressure, only
cause sclerosis of the afferent arteriole.)
* Atherosclerosis of intrarenal arteries is common in diabetics and rare in non-diabetics; it is not the
major problem.
Glomerular lesions
Always present:
1. Thickening of the glomerular basement membrane because of increased production of GBM
(sometimes called "diffuse glomerulosclerosis").
2. Increased amounts of mesangial matrix (also sometimes called "diffuse glomerulosclerosis").
Increased number of mesangial cells in the early lesion, later decreased as the entire glomerulus
is replaced by matrix ("hyalinization" of the glomerulus.)
* 3. The GBM, mesangial matrix, and tubular basement membranes (also thick) bind
albumin
and other proteins non-specifically ("all that sticky sugar....")
* These three features, together, are pathognomonic of diabetes mellitus (but you probably knew
already....) They occur separately in other diseases.
Often present:
Nodular glomerulosclerosis or (nodular) Kimmelstiel-Wilson disease. Big balls of
GBM-mesangial matrix material in the glomerular tufts. Highly characteristic of diabetes
or FSGS.
{08892} KW disease; note balls of hyaline, and thick GBM
(i.e., you can actually tell where it is)
Sometimes present:
* "Fibrin caps" ("exudative lesion", "hyperfiltration lesion") -- hyaline crescents on a glomerular tuft
* "Capsular drops" -- hyaline material on the inside surface of Bowman's capsule (highly characteristic
of diabetes.)
Clinically, patients have albuminuria (rarely heavy proteinuria), then renal failure (probably due to the
mesangium crunching the glomerular capillaries).
The etiology of diabetic glomerulopathy is complex and poorly-understood. Intrarenal fluid dynamics
are involved. We don't even know why the kidneys enlarge in
diabetics (NEJM 324: 1662, 1991, still good).
Tight control of blood glucose does seem to benefit these patients, and reduces the hyperfiltration
response to amino acids (NEJM 324: 1629, 1991). Patients are now put on ACE-inhibitors and
protein-restricted to prevent progression of the renal disease. (Yes, it can regress
de to therapy: NEJM 348: 2285, 2003).
* Ace-inhibitor plus a calcium channel blocker works marvellously
to prevent diabetic kidney disease: NEJM 351: 1941, 2004.
Other renal lesions in diabetes:
* Thick tubular basement membranes (not a health problem).
* Fatty change of tubular cells (systemic lipid disturbance, not a health problem).
* Glycogen in proximal tubular cells (Armanni-Ebstein lesion, a sign of heavy glycosuria, not itself a
health problem).
{46306} Armanni-Ebstein; lots of glycogen in the
tubular cells
Kidney infections (gram-negative bacilli causing infection of renal pelvis in pyelonephritis,
staphylococci causing cortical infections, candida infections, etc.)
Renal papillary necrosis -- just like it sounds. (* "Baby Robbins" misnames it
"necrotizing papillitis".
The lesion is seen in diabetes, obstruction, sicklers, Wegener's, or abuse of the analgesic
phenacetin.)
{49306} pyelonephritis and papillary necrosis in a diabetic
EYES: Diabetes is the commonest cause of blindness before old age in the US.
Review: Lancet 350: 197, 1998.
Cataracts: a variety of types, including some clearly caused by sorbitol deposition (proof Proc. Nat.
Acad. Sci. 9: 2780, 1995).
Glaucoma: reason for its being more common with diabetes is uncertain.
Diabetic retinopathy: the most serious diabetic eye problem
Nonproliferative phase (NEJM 322: 978, 1990)
Edema, protein exudates, hemorrhages, microinfarcts ("cotton-wool patches") all indicate vascular
problems
Microaneurysms (the first change,
and highly characteristic of diabetes): ballooning of capillaries where perhaps a
pericyte has come off.
{09365} diabetic retinopathy; hemorrhages and exudates
Proliferative phase: new vessels grow, eventually invading vitreous humor, with hemorrhage,
granulation tissue, fibrosis, retinal detachment. These patients get photocoagulation.
{09366} proliferative retinopathy
The molecular biology remains puzzling.
Sudden normalization of a poorly-controlled diabetic's glucose can accelerate proliferative
retinopathy (Arch. Ophth. 116: 874, 1998).
PERIPHERAL NERVES (morphology: Diabetes 46 S 2: S 50, 1997)
Manifests as symmetrical sensory loss, sometimes with uncomfortable paresthesias,
and as autonomic disturbances such as diarrhea
(Am. J. Gastro. 94: 2165, 1999), bladder
problems, orthostatic hypotension, impotence. Less often a mononeuropathy, perhaps due to
infarction of a nerve.
Axons are lost, and Schwann cells also take a beating.
{48174} Charcot's neuropathic joint changes
Probable chemistry: increased availability of glucose for polyol pathway results in more sorbitol.
* Aldose reductase produces polyols
that are linked to the late complications
in nerve and kidney. Inhibitors were not a great
success for the neuropathy, but different aldose reductase alleles confer
susceptibility to or protection from the glomerulopathy.
(Diabetes 46: 287, 1997.)
The most interesting new work in diabetic neuropathy focuses on
the ability of ACE inhibitors to stop the progression independent of
effects on blood pressure (NEJM 345: 851, 2001) PANCREATIC ISLETS -- except when the islands are actually destroyed, the relationship of
morphologic lesions to the abnormal metabolic state is usually obscure.
In Type I diabetes mellitus, early cases show lymphocytic infiltration ("insulitis"). Later cases show
destruction of beta cells and even whole islets.
Degranulated and/or glycogen-filled beta cells have also been described.
In Type II, the islets most often look normal.
In either Type I or Type II, there may be hyalinization of the islets with collagen and/or amyloid. The
latter is beta-pleated amylin, that new islet hormone.
It is common in Type II diabetes,
it may precede the overt disease, and it now appears that its massive accumulation in islets
of type II diabetes does impair insulin production, and possibly is toxic to the beta-cells (Nature 368:
756, 1994). Confirmation: J. Clin. Endo. Metab. 89: 3629, 2004.
{08084} hyalinized pancreatic islets, type II diabetes
Babies of diabetic mothers have hyperplastic islets (because of all that glucose),
and they are infiltrated
with lymphocytes and eosinophils (mysterious.)
OTHER PROBLEMS FOR DIABETICS
Infections (bacterial and fungal)
Why diabetics get more infections is still poorly-understood. Candida may thrive on the glucose,
hyperglycemia slows down polys, poor circulation keeps the body from fighting infection, etc., etc.).
{48090} diabetic abscess
Gallstones (made of cholesterol; nobody knows why these
are more common in diabetics, but the average gallbladder volume
is much higher in non-insulin-dependent diabetics, perhaps
promoting stasis and nidation: Dig. Dis. Sci. 43: 344, 1998.)
Altered platelet function (significance?)
Complications of pregnancy -- all the common problems are commoner in diabetic mothers, and babies
are bigger (partly the hyperglycemia, probably partly some growth factor or other: Br. J. Ob. Gyn. 103:
427, 1996) and at extra risk for a variety of birth defects (all of which
seem to be preventable by euglycemia through pregnancy).
Diabetic xanthomas (yellow skin bumps -- pseudotumors made of lipid-laden macrophages),
necrobiosis (focal necrosis of the dermis), and many other skin abnormalities
{12214} necrobiosis lipoidica diabeticorum
Hepatic fatty change, even in sober diabetics ("non-alcoholic
steatohepatitis", which runs the gamut through neutrophisl and Mallory's
hyaline to micronodular cirrhosis). Probably this has to do with Syndrome X. Stay tuned.
* Scleredema -- pseudosclerodermatous changes over the back and shoulders caused
by accumulation of glycosaminoglycans. This may be a marker for longstanding poor control.
* "Diabetic dermopathy" is purple-brown patches on the shins (less often, the upper legs and/or forearms)
which may grow to coalesce. This supposedly has something to do with the microangiopathy
and may be seen in other situations with vascular insufficiency.
Chronic hyperglycemia results in non-enzymatic glycosylation of many body proteins.
Hemoglobin A1c is glycosylated hemoglobin that can be measured in the blood to assess the quality
of diabetic control (though, of course, home blood glucose testing several times a day by a highly
motivated patient is even better....)
The literature is suddenly exploding with talk about "advanced glycosylation (glycation) products", i.e.,
proteins that have undergone a series of reactions with glucose. For one thing, at least some human
cells have a surface receptor for these products, which then activate genes in blood vessels (Proc. Nat.
Acad. Sci. 91: 8807, 1994) and glomeruli (Proc. Nat. Acad. Sci. 91: 9519, 1994; Diabetes 44: 824,
1995).
Hemoglobin A1c is an obvious choice for a diabetes screening device, and has been studied
as such (JAMA
276: 1264, 1996); it's still not in common use.
Much more about the laboratory diagnosis of diabetes and hypoglycemia is available from your lecturer.
Phone me when you're on rotations if I can help you with a diabetes-related problem. Some current
articles:
J. Clin. Endo. Metab. 85: 1584, 2000.
Glycogen synthetase is deficient in diabetic muscle, but contrary
to older reports, this is probably the result rather than the cause
of type II diabetes.
NEJM 346: 393, 2002. Exercise can actually forestall the development of type II diabetes; apparently,
the more, the better; supports many other studies, and better than metformin.
Br. Med. J. 318: 1169, 1999. the current pop claim that
hemophilus influenzae B vaccine causes diabetes in children doesn't hold up.
NEJM 350: 1398, 2004. The current claims that immunization causes
diabetes are examined in a massive Danish
study. Intense scrutiny of kids who did and did not get each of the
common vaccines shows no apparently difference in the risk for diabetes. JAMA 279: 669, 1998. Regular brisk exercise is still
a great way to improve or maintain insulin sensitivity.
Diabetes 40: 161, 1991. Speculative article with an archeological twist, concerning selection for type
II diabetes genes in hunter-gatherer populations faced with episodic famine; intended to "explain" high
prevalence in descendants of the first Amerindians.
Science News 140: 11, 1991. WHO recognizes "malnutrition related diabetes", a diabetes variant
beginning during the teenaged years; victims are mostly survivors of childhood malnutrition who also
eat lots of cassava, a cyanide-rich food.
NEJM 330: 962, 1994. A mitochondrial gene mutation that produces diabetes and deafness. Nobody
knows how. The original big paper.
NEJM 353: 704, 1995. Neonatal diabetes -- I cared for "Baby Sweetwater" s a medical student.
Ann. Int. Med. 109: 639, 1988. Screening healthy adults for NIDDM is a big waste.
Br. Med. J. 308: 611 & 1639, 1994. Looking for diabetes.
Br. Med. J. 308: 632, 1994. Ad campaign about symptoms of diabetes.
Br. Med. J. 303: 260, 1991. "Brittle diabetes" in today's world probably means noncompliance.
Diabetes 46: 688, 1997. Whatever
that thing in the islands that binds sulfonylureas does,
a mutation seems to cause morbid obesity and diabetes.
Diabetes 36: 434, 1987. Even running once around the track improves insulin sensitivity.
NEJM 309: 44, 1983. Maybe refined sugar's not so bad for diabetics....
Lancet 2: 122, 1984. Forbidding refined sugar for diabetics is superstitious....
Am. J. Clin. Nutr. 78: 858-S, 2003. Organized medicine has finally dropped
the goofy requirement that diabetics shun all refined sugar and limit their intake
of natural simple sugars. The "glycemic index" for various foods also seems to be a myth
(i.e., the old story about "simple sugars are absorbed faster" isn't so -- we've
actually know this for decades. Type I's: Adjust your insulin by how much carbohydrate you
plan to eat. Type II's: Keep on that calorie-restricted diet.
NEJM 315: 224, 1986. Fiber's good, simple sugars seem OK, real knowledge is scanty.
JAMA 256: 3241, 1986. Yeah, some sucrose is permissible, fructose is better.
Lancet 361: 2005, 2003. Simvastatin slows atherosclerosis
in diabetics and helps even if cholesterol isn't that high.
JAMA 282: 750, 1999. Diabetics may not realize their blood glucose
levels are too low to permit safe driving, and may drive even if they know.
Am. J. Clin. Nut. 54: 846, 1991. The arcane "glycemic index" for food, a measure of how badly a
particular dietary item will throw a diabetic's glucose and maybe cholesterol out of kilter.
JAMA 271: 1421, 1994. High-carbohydrate diets for NIDDM tend to throw them out of kilter.
High-monounsaturated diets are better.
Br. Med. J. 307: 292, 1993. Managing diabetic Moslems during Ramadan.
JAMA 257: 81, 1987. Diabetics in control can do most any job.
JAMA 272: 305, 1994. All about the many LDL's, including those in diabetes.
Br. Med. J. 299: 591, 1989. Being on insulin should not raise your driver's insurance premium.
Lancet 1: 599, 1985. Cyclosporine as immunosuppressive treatment of Type I diabetes.
Lancet 363: 925, 2004. A "pop" claim that nicotinamide prevented
type I diabetes in kids forced the British to do a prosepctive study;
despite some support in an animal model, it didn't work for humans.
JAMA 281: 2005, 1999. review of getting type II diabetes under control.
NEJM 333: 381, 1995. Beta-3 adrenergic receptor, obesity, and Finnish familial obesity.
Lancet 336: 402, 1990. First successful islet transplant in humans.
Diabetes 50: 47, 2001. When you take out the pancreas to treat
intractable pain from chronic pancreatitis, it's good to autotransplant
islands into the liver to prevent diabetes!
J. Immuno. 163: 1178, 1999. giving the epitope to suppress the
autoimmune destruction of the pancreas.
Nat. Med. 6: 278, 2000: Ixion Biotech claims to reverse
diabetes in mice using stem cells. Today's politics in the US are forcing
this work to go overseas. Stay tuned.
Lancet 364: 203, 2004. Update on stem cells for type I diabetes.
The mouse models are encouraging.
Science 260: 1942, 1993 transgenic mice with islands that over-express α interferon get
autoimmune diabetes.
Lancet 343: 95, 1994. Caring for non-insulin-dependent diabetes.
Lancet 346: 157, 1995. The pregnant type I diabetic.
NEJM 322: 1028, 1998. gastric motility disturbances.
BMJ 319: 83, 1999. Self-monitoring is a big help for glycemic control,
but even most type I diabetics don't do it.
Am. J. Med. 111: 1, 2001. Kaiser Permanente finds that the more you
monitor yourself, the better your control, and that this is good.
Postgrad. Med. 109: 41, 2001. Present and future technology
for glucose monitoring. Some talk about "the mechanical islet".
Crit. Care Med. 29: 1062, 2001. In glucose oxidase-based test strips
that measure amperage instead of color changes, high oxygen tensions. above 100 torr.
in the blood can give a false-low glucose level.
Proc. Nat. Acad. Sci. 90: 5843, 1993. Long-term reversal of the diabetic state by implantation of islets
immunoprotected by alginate poly-amino acid capsules. I hope the heck this works.
Lancet 342: 129, 1993. Tight glucose control seems to prevent many complications, especially for type
I.
Arch. Int. Med. 163: 101, 2003. Underclass diabetics have a much higher
rate of hospitalization for diabetes even when they have good access to outpatient
health care.
Am. J. Psych 150: 1114, 1993. The more you and your family get emotional
in criticizing each other, the better your diabetic control. Weird article.
NEJM 338: 867, 1998. Metformin (less gluconeogenesis) and troglitazone (more insulin
sensitivity) work together for the great benefit of the diabetic.
J. Clin. Endo. Metab. 88: 2412, 2003. The thiazolidinediones
are a new class of insulin sensitizer that work by binding to
the nuclear receptor peroxisome proliferator-activated receptor gamma (PPAR-gamma).
This seems to target the metabolic syndrome ("syndrome x").
J. Geront. 48: M-117, 1993. Older type II diabetics think more clearly if their blood glucose is better
controlled.
Lancet 335: 595 & 652, 1990. Appallingly, ketoacidosis is still sometimes missed.
South. Med. J. 91: 151, 1998. Appallingly, ketoacidosis and
other deadly manifestations of diabetes still get missed, even in the US.
JAMA 269: 619, 1993. The cops take away your insulin when you go to jail, and you can die.
Diabetes Care 13: 188 ff., 1990. Easy-to-read articles on the molecular biology of diabetes.
JAMA 271: 275, 1994. ACE inhibitors to slow the progression of diabetic nephropathy.
NEJM 330: 15, 1994. The above must be working, we have a lot less diabetic nephropathy than we
used to.
Am. J. Psych. 147: 1275, 1990. Emotions and diabetes, a complex story.
NEJM 331: 854, 1994. All about the diabetic foot.
MMWR 40: 229, 1991. Diabetes is expensive!.
JAMA 292: 1711, 2004. Adults who can't read very well
don't take very good care of their diabetes. The authors suggest
working on reading skills; perhaps instead adult illiteracy is symptomatic
of a deeper indifference.
Br. Med. J. 308: 1208, 1994. "Integrated care" by diabetes teams.
HYPOGLYCEMIA -- worth mentioning concurrently with diabetes mellitus
Hypoglycemic coma: When patients come to the emergency room in coma, you'll probably give them
50 gm of glucose by vein just after you draw their blood work. This brings around the known diabetics
who've overdosed on their insulin ("insulin shock"), and the undiagnosed type II diabetics who are sick
(post-prandially) from "too much insulin too late". Both situations are very common.
Fasting hypoglycemia is always worrisome. You need to rule out beta cell tumor ("insulinoma" --
the most common islet cell tumor), factitious illness (illicit / murderous administration of insulin and/or oral
hypoglycemic agents), and a few rarities.
Post-prandial (maybe 2-4 hours after eating)
hypoglycemia in non-diabetics is seldom serious. Symptomatic "hypoglycemia" in
non-diabetics, a fad diagnosis in some circles a few years ago, actually results from an abnormally brisk
epinephrine response to a falling plasma glucose (JAMA 251: 612, 1984; Soc. Sci. Med. 22: 599,
1986).
The popularity of this diagnosis enabled the San Francisco "Twinkie" murderer to escape punishment.
However, the familiar queezy syndrome probably has nothing to do with refined sugar, which in turn is
probably not the cause of all social evils....
The medical name for the familiar queezy syndrome is "idiopathic postprandial syndrome". Naturally,
it's worst when biopsychosocial stresses are greater. Keep raisins in your pocket on your rotations. "Even when intake exceeds typical dietary levels, neither dietary sucrose nor aspartame affects children's
behavior." The word from NEJM 330: 301 & 335, 1994 (big Vanderbilt study using kids whose
parents thought they were "sensitive to sugar" or whatever).
PANCREATIC ISLET CELL TUMORS: Not-to-be-overlooked causes of striking clinical syndromes.
Complex diagnostic and surgical problems. (Clinical reviews -- Surgery 99: 671, 1986; Curr. Prob.
Surg. 27: 301, 1990; new hope through chemotherapy NEJM 326: 519, 1992.)
{24576} islet cell adenoma
Once considered "exotic", a Toyko study recently found an endocrine adenoma in 3% of patients
coming to autopsy for any reason (Dig. Dis. Sci. 36: 933, 1991).
The tumors are usually slow-growing, even when malignant (10% of total, higher for glucagonomas,
gastrinomas, ACTH-omas and somatostatinomas). See Radiology 190: 59, 1994. You need to have
a high index of suspicion to detect these, and all other endocrine diseases.
These tumors make one or more of the following: glucagon, insulin, gastrin, somatostatin, vasoactive
intestinal polypeptide (VIP), pancreatic polypeptide (PP), * neurotensin, and * neuron-specific enolase.
Check your patient's serum -- elevated levels of one or more of these should prompt a search for an
islet-oma (angiogram, CT scan, maybe surgery.
The surrounding pancreatic islets may show hyperplasia, * nesidioblastosis, etc. * Future surgical
pathologists study Arch. Path. Lab. Med. 125: 1344, 2001).
Metastatic spread remains the only reliable criterion for malignancy in these tumors.
Beta cell tumors ("insulinomas"): the most common islet cell tumors (* but their incidence is only
about 1 in 1,000,000 people)
Look for Whipple's triad:
Often, patients become massively obese.
The etiology of these tumors is obscure; there's not even an important genetic syndrome.
Of beta-cell tumor patients, 70% have a solitary adenoma, while the rest have either hyperplasia of
many islands (* "nesidioblastosis"), or a beta cell carcinoma
(the spectrum to malignancy: Cancer 104: 264, 2005).
* "Leucine-induced hypoglycemia" often heralds an insulinoma.
* Future surgeons: If the plasma insulin isn't down eight minutes
after you remove the tumor, you didn't get it all. Surgery 132: 937, 2002.
* NOTE: Nesidioblastosis is a rare, poorly-understood cause of hypoglycemia in infancy. The islands
and/or periductal tissue is packed with immature islet cells. Adults can get it too (Arch. Surg. 129 329,
1994), nobody knows how.
* Speaking of nesidioblastosis... Following the newly-popular Roux-en-Y
gastric bypass surgeries, patients typically develop some hyperplasia of the islets,
relieving any type II diabetes that may be present and sometimes even resulting in
hypoglycemia (once attributed to dumping syndrome). Exactly what gut hormones cause
this, and how, is being worked out. See NEJM 353: 249, 2005.
* NOTE: Many mesotheliomas and retroperitoneal fibrosarcomas, and occasionally other tumors,
produce an insulin-like activity (probably somatomedin, but it varies).
PITFALL: The insulin produced by these tumors is often a bit abnormal, and
a lab assay can miss it: J. Clin. Endo. Metab. 88: 1464, 2003;
Lancet 363: 363, 2004.
NOTE: Be alert for factitious hyperinsulinism! To differentiate sly insulin administration from a
beta-cell tumor, start by measuring plasma C-protein. To rule out oral hypoglycemic drug
administration, check serum drug levels. Gastrinomas (Zollinger-Ellison Syndrome; "G-cell tumors", etc.;
pathologist's review Cancer 68: 1329, 1991.)
An especially troublesome syndrome of multiple bleeding ulcers and diarrhea. The majority of
gastrinomas are low-grade malignancies. The only way to tell is whether it has metastasized.
High basal acid secretion plus a marked increased in serum gastrin levels in response to secretin
administration strongly suggests gastrinoma (Am. J. Med. 80: 11, 1986.
Screening asymptomatic persons merely turns up lots of older people with achlorhydria (Br. J. Surg. 77:
1, 1990).
Removal of all the tumor, if possible, is curative. Otherwise, these patients used to require total
gastrectomy, but now they respond well to H2-blockers. Somatostatin analogues (notably "Octreotide",
available from your pharmacist) are an effective alternative.
{09279} gastrinoma (to prove it, we'd need to
do an immunoperoxidase stain that stains gastrin brown and everything
else white)
Glucagonomas ("alpha-two cell tumors"):
These produce mild diabetes, sore tongue, and necrolytic migratory erythema
(death of the layer of cells 3/4 of the way up the epidermis). Don't miss this diagnosis
(and people do: West. J. Med. 144: 746, 1986) -- even when metastatic, it responds well to somatostatin
and its analogues (Acta Med. Scand. 218: 245, 1985; NEJM 314: 1686, 1986; Ann. Int. Med. 110: 35,
1989), and to oral zinc therapy. Update Am. J. Med. Sci. 321: 306, 2001;
Gastroenterology 116: 1286, 1999.
Multiple Endocrine Neoplasia Syndromes ("MEN", formerly "MEA", adenomas): some or all of the
following in same family
Wermer's MEN I: pituitary adenoma, parathyroid adenoma, pancreatic endocrine tumors (
most often gastrinoma / Zollinger-Ellison; less often insuiinoma, others)
Sipple's MEN II(a): Parathyroid adenoma, pheochromocytoma, medullary carcinoma of the thyroid
MEN IIb/III: Medullary carcinoma of the thyroid, pheochromocytoma, mucosal neuromas, Marfanoid
habitus
More about these later....
Rarities:
Delta cell tumors ("somatostatinomas"): diabetes, diarrhea, gallstones, etc. Review: J. Surg. Onc. 43:
259, 1990.
Tumors secreting vasoactive intestinal polypeptide ("VIPomas"; "Verner-Morrison syndrome"): pancreatic cholera (horrible diarrhea),
loss of potassium, achlorhydria -- excellent response to somatostatins.
PP-omas ("P-cell tumors"): no syndrome despite huge amounts of pancreatic polypeptide (Cancer 57:
129, 1986). * Atropine suppression test: NEJM: 315: 287, 1986).
Even if you open up a practice
Years from now
Those among us who are diabetic
--Ron Charach, M.D.
 Cystic fibrosis
Cystic fibrosis
Photo and mini-review
Brown U.
 Hemorrhagic pancreatitis
Hemorrhagic pancreatitis
Urbana Atlas of Pathology
{08342} enzymatic fat necrosis, microscopic;
left upper corner
{08348} enzymatic fat necrosis, microscopic
Pancreatic pseudocyst
Spectacular x-ray
Brazilian Medical Students
Also helpful for spotting cancer: glands abutting smooth muscle, or perineural
invasion, weird nucleoli.
{08853} chronic pancreatitis; extensive scarring
{20279} chronic pancreatitis in an alcoholic, nice protein plug
{46283} chronic pancreatitis with calculi
{49230} burned out alcoholic chronic pancreatitis, with calculi;
the tube along the bottom is the splenic artery, twisting in and out
Chronic alcoholic
Chronic obstructive Lobules unevenly scarred
All lobules in an area involved equally Protein plugs in small ducts
Few or no protein plugs Perineural chronic inflammation
Normal nerves
* Truly hardcore future pathologists will want to read about
MUC4 as a stainable marker for pancreatic neoplasia, especially
as it grows nastier: Am. J. Clin. Path. 117:
791, 2002.
{26003} adenocarcinoma of pancreas; mucin-producer
(pale apical cytoplasm, sharp borders)
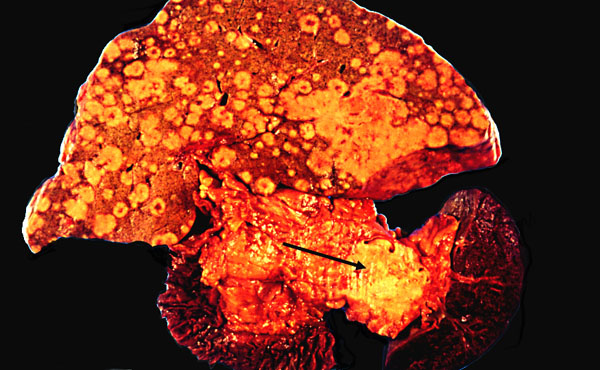 Cancer of the pancreas
Cancer of the pancreas
Primary and liver metastases
KU Collection
 Risk
factors include (1) smoking (3x the normal risk), (2) exposure to chemicals (the disease is
supposedly more common among both chemists and garage workers), (3) hereditary
pancreatitis (huge risk), and (4) Some of the anti-oncogene deletion syndromes.
Risk
factors include (1) smoking (3x the normal risk), (2) exposure to chemicals (the disease is
supposedly more common among both chemists and garage workers), (3) hereditary
pancreatitis (huge risk), and (4) Some of the anti-oncogene deletion syndromes.
15% body
5% tail
20% too late to tell
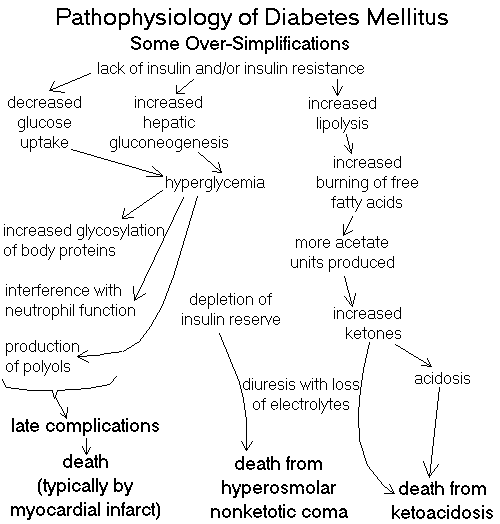
 "Diabetes" literally means "siphon", because of the osmotic
diuresis produced by the glycosuria. This was known all-too-well to Hippocrates,
who may have named it.
"Diabetes" literally means "siphon", because of the osmotic
diuresis produced by the glycosuria. This was known all-too-well to Hippocrates,
who may have named it.
The new system recommended by the WHO and the American Diabetes Association
(Br. Med. J. 317: 359, 1998):
The fasting criteria are down from 140 mg/dL (Am. Fam. Phys. 58:
1355, 1998). It identifies people at risk for
health problems from hyperglycemia.
Type I: autoimmune and idiopathic situations in which beta cell function is
essentially gone, and there is an absolute insulin lack;
Insulitis
Type I diabetes
WebPath photo
* All the recent stuff is from obvious
"independent thinkers" (ignoring what we know of immunology: Food & Chem. Tox. 42: 707, 2004)
and studies that invited recall bias (Ann. Nutr. Metab. 47: 267, 2003).
In the non-obese diabetic (NOD) mouse, there is a specific defect
in a group of T-suppressor cells. * A possible ligand to
enhance these cells in mice and humans: Nat. Med. 7: 1052, 2001.
Why these usually do not declare themselves at birth remains a mystery.
Perhaps all that would do so have been strongly selected-against.
Expect LOTS more to be discovered.
There is probably even more type II diabetes in the poor nations today
than in the U.S.
Blacks 25%
Whites 17%
* A novel new study found that diabetics and obese folks
have lost many of their
subsarcolemmal mitochondria, and those that remain do not function well
(Diabetes 54: 8, 2005).
Of course acanthosis nigricans is a darkening and thickening of
the epidermis in the armpits and groin. When not part of a
paraneoplastic syndrome, it is usually a marker for insulin resistance
of some cause; however the underlying basic biology
remains elusive.
Good glycemic control does help the accelerated
atherosclerosis, confirming the idea that it's due largely to
the accumulation of advanced glycation products that cause collagen production.
{48076} diabetic gangrene
{48022} diabetic ulcer
{48023} diabetic ulcer
{48150} diabetic ulcer
)window.location='http://edcenter.med.cornell.edu/Pathology_Images/329.gif') Diabetic gangrene
Diabetic gangrene
Cornell
{17159} diabetes with hyalinized arteriole
{16789} diabetic glomerulosclerosis, electron micrograph (thick GBM)
{16790} diabetic glomerulosclerosis, electron micrograph (thick GBM)
{16791} diabetic glomerulosclerosis, H&E
{16792} diabetic glomerulosclerosis, PAS; nice
capsular drop too
{16793} diabetic glomerulosclerosis, H&E
{08893} Kimmelstiel-Wilson diabetic nodular glomerulosclerosis;
H&E
{08895} Kimmelstiel-Wilson diabetic nodular glomerulosclerosis, PAS
{09877} Kimmelstiel-Wilson diabetic nodular glomerulosclerosis
{17158} Kimmelstiel-Wilson diabetic nodular glomerulosclerosis
{17171} end-stage diabetic glomerulosclerosis
{22036} diabetic retinopathy; microaneurysms
{22039} diabetic retinopathy
{22042} diabetic retinopathy, notice the hemorrhages
{22045} diabetic retinopathy -- bleed
{22904} microaneurysm
{23156} cotton wool patches
{23180} diabetic retinopathy
{23183} diabetic retinopathy
{22895} proliferative retinopathy
{22901} proliferative retinopathy -- "scar contracts"
and tears off the retina
Amyloid islet
Type II diabetes
WebPath photo
{48091} diabetic abscess
Diabetic skin
Lecture notes and
some great photos
* One team found that nonenzymatic glycosylation actually altered
keratinocyte surface receptors so they could not take up glucose. This may
have something to do with the poor epidermal healing and some of the other
changes (Diabetes 50: 1627, 2001).
We pathologists like to see you order a hemoglobin A1c on stable patients twice
a year, with the target of a final HgbA1c value <7%. Arch. Path. Lab. Med. 215:
191, 2001.
 * ADDITIONAL INFORMATION ABOUT DIABETES
* ADDITIONAL INFORMATION ABOUT DIABETES
It takes only a few seconds to make up a lie. It takes years [and in this
case, millions of dollars] to refute it. And even then, people still adopt
their "most cherished beliefs" on emotion. -- Ed.
 Student Doctor
Student Doctor
Alex Yartsev
"Pathology Outlines"
on diabetes MD
 Pancreatic islet cell tumor
Pancreatic islet cell tumor
Great photos
Pittsburgh Pathology Cases
{49239} islet cell adenoma -- top center
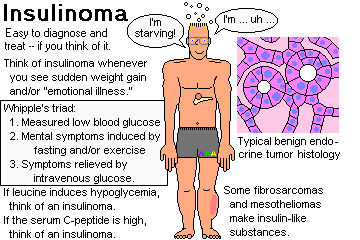
{09282} gastrinoma
Glucagonoma
Pittsburgh Pathology Cases

On Harley Street
No patient will come in with complaints
about his pancreas:
"I think it's my pancreas, Doc!"
-- unless he's a fellow professional
also educated
out of his natural mind; few patients
will be alarmed by the word -- how unlike
"the heart"
a word that means "the biscuit"
to the best of us.
when you trundle in
thin and yellow, depressed,
for abdominal films,
you too will have forgotten
your pancreas; and the news "It's cancer
of the pancreas" will hit
like an old family secret you knew all along;
"I'm sorry, but it's cancer
of the sweetbread!"
"Not the sweetbread!" -- "Yes,
and, with proper medical management
early surgery
and a very rigid diet
you can look forward to at least
another three months"; when the pancreas goes
it goes.
whom the pancreas torments
by degrees
cannot describe that Familiar; even a poet
is at a loss for a metaphor;
nothing short of a surgical exploration
will unearth
the thick spongy worm
buried deep in the viscera
silent behind its curtain of peritoneum;
-- with a head, a body,
and a tail,
using the man's face.
NEJM 301: 508 (1979)
 * SLICE OF LIFE REVIEW
* SLICE OF LIFE REVIEW
08093 islet, normal
11748 pancreas, normal
14886 pancreas, normal
14886 pancreas, normal
14887 pancreas, normal
14888 pancreas, duct & islets
14889 pancreas, duct & islets
14890 pancreas, serous acini & islet
14891 pancreas, serous acini & islet
14892 pancreas, exocrine and endocrine
14893 zymogen cells, normal pancreas
14894 islet of Langerhans
14895 islet of Langerhans
14896 pancreas, zymogen granules
14897 pancreas, zymogen granules
14898 pancreas, central acinar cells
14899 pancreas, central acinar cells
15274 pancreas, normal
15275 pancreas, normal
15276 islet of Langerhans, #65
15277 pancreas, intercalated duct
15278 pancreas, centroacinar cell and duct
15279 islet of Langerhans, #65
15280 ?pancreas, ct in duct??
15793 pancreas, normal
20879 pancreas
20880 intercalated duct, pancreas
20881 acinar cell, pancreas
20882 islets of langerhans, pancreas
20883 pancreas, centroacinar cell
20884 pancreas
20885 islet of Langerhans
20886 pancreas, interlobar duct
20887 pancreas, interlobar duct
25019 pancreas, normal
25020 pancreas, normal

25021 pancreas, normal
25794 columnar cells, pancreatic epithelium - norma
25973 pancreas, normal
25988 pancreas, normal
25991 acinar cells, normal pancreas


| Visitors to www.pathguy.com reset Jan. 30, 2005: |
Ed says, "This world would be a sorry place if
people like me who call ourselves Christians
didn't try to act as good as
other
good people
."
Prayer Request
 Teaching Pathology
Teaching Pathology
PathMax -- Shawn E. Cowper MD's
pathology education links
Ed's Autopsy Page
Notes for Good Lecturers
Small Group Teaching
Socratic
Teaching
Preventing "F"'s
Classroom Control
"I Hate Histology!"
Ed's Physiology Challenge
Pathology Identification
Keys ("Kansas City Field Guide to Pathology")
Ed's Basic Science
Trivia Quiz -- have a chuckle!
Rudolf
Virchow on Pathology Education -- humor
Curriculum Position Paper -- humor
The Pathology Blues
 Ed's Pathology Review for USMLE I
Ed's Pathology Review for USMLE I
![]()
![]()
 | Pathological Chess |
 |
Taser Video 83.4 MB 7:26 min |

 Pancreas
Pancreas
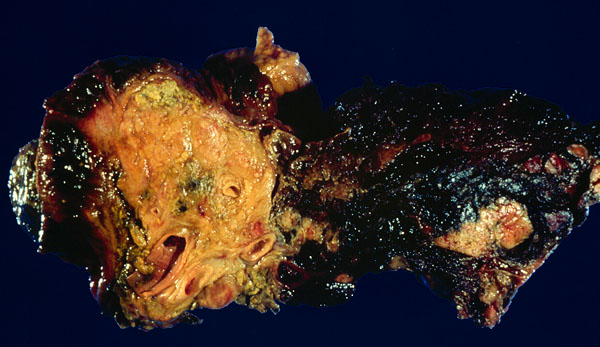 Hemorrhagic pancreatitis
Hemorrhagic pancreatitis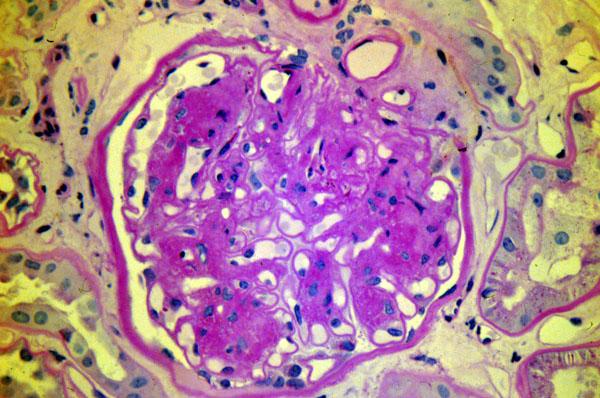 Nodular glomerulosclerosis
Nodular glomerulosclerosis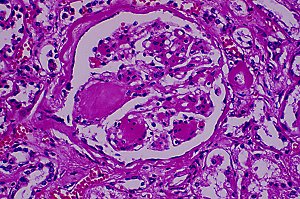 Nodular glomerulosclerosis
Nodular glomerulosclerosis